
成果简介
这项研究展示了在发现高熵合金(HEA)电催化剂方面取得的显著进展。通过利用机器学习(ML)来简化密度泛函理论(DFT)计算的需求,该研究将吸附位点的相似性纳入图神经网络,显示出训练速度提高了23%。这一方法有效减少了使用计算密集型DFT计算生成数据集的工作量。
研究成功验证并实验鉴定了HEA电催化剂在氧还原反应(ORR)中的性能,表现优于传统的Pt/C催化剂。展望未来,这种方法有望广泛应用于更广泛的材料系列,如高熵氧化物和氮化物。其潜力在于能够彻底改变催化剂的发现过程,从而更快、更有效地探索复杂的化学空间,并最终促进能源转换技术和可持续化学过程的发展。
多相催化的重要性与挑战
为进一步深入研究 HEA 电催化剂,我们需探索广阔的化学空间,涵盖多种元素组合和原子构型。通过仅对部分表面性质进行密度泛函理论 (DFT) 计算,并基于此子集训练机器学习 (ML) 模型替代昂贵的高性能计算,有望显著降低计算成本,从而高效筛选具有理想催化特性的 HEA 材料。
HEA 电催化剂的计算流程
为加快模型训练速度,我们提出了新的输入特征,以减少达到目标精度所需的数据点数量。基于相似属性的假设,我们引入了一种相似性度量,认为通过加入该度量,可以基于已知结构的能量更准确地预测未计算结构的能量。具体而言,我们测量某一化学位点(例如催化剂表面的原子)与其他位点之间的相似性。在机器学习 (ML) 模型训练过程中,我们将这一相似性度量作为特征,以便在更少数据点的情况下达到给定的测试精度。为此,我们开发了基于增强晶体图卷积神经网络 (CGConv) 的 ML 框架,如图 1 所示。

为便于与先前开发的方法进行性能比较,我们将研究重点放在了五元材料系列的铁钴镍铱钌 (FeCoNiIrRu) 合金上。训练数据集和预测数据集均为随机选择。首先,我们对随机选择的 987 个 FeCoNiIrRu 3×4 HEA 表面进行了密度泛函理论 (DFT) 计算,计算了 OH* 和 O* 的吸附能。
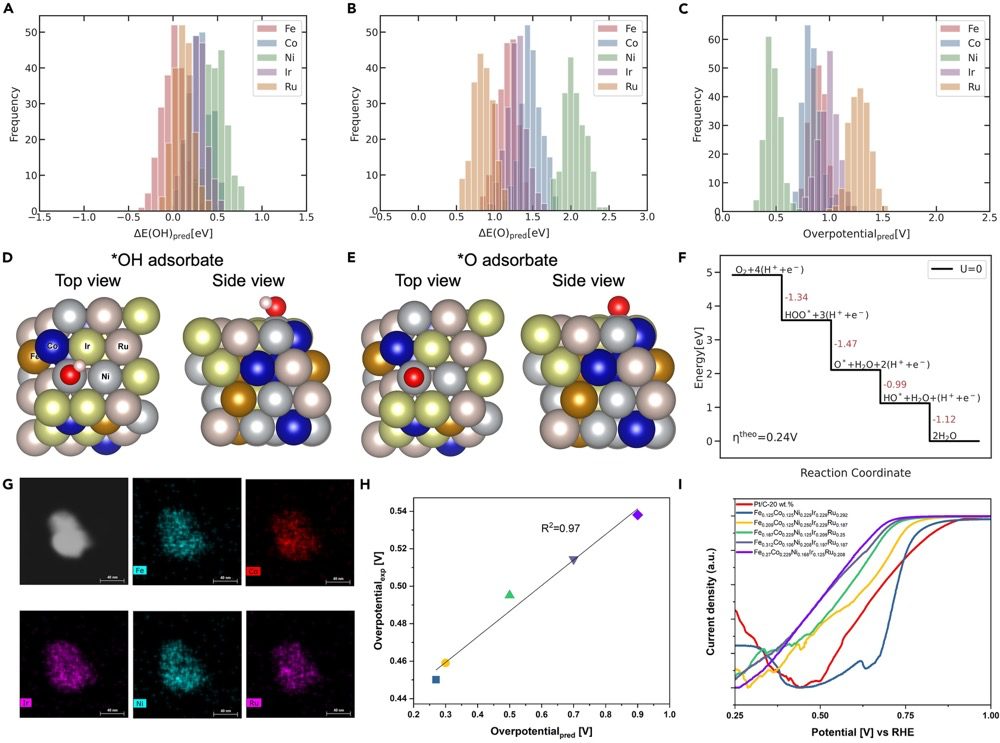
随后,我们对随机选择的 1,000 个 HEA 表面进行了吸附能预测,并基于吸附能计算得出过电位,以评估其氧还原反应 (ORR) 活性。各元素的 O* 吸附能分布较为分散,说明与吸附位点对应的元素对 O* 吸附能有显著影响。相比之下,OH* 吸附能的分布范围较窄。DFT 数据集中的最佳候选者为 Fe0.188Co0.125Ni0.125Ir0.333Ru0.229,其中吸附物位于 Ni 的顶位。

相似性和多样化:使用富士通量子启发数字退火实施
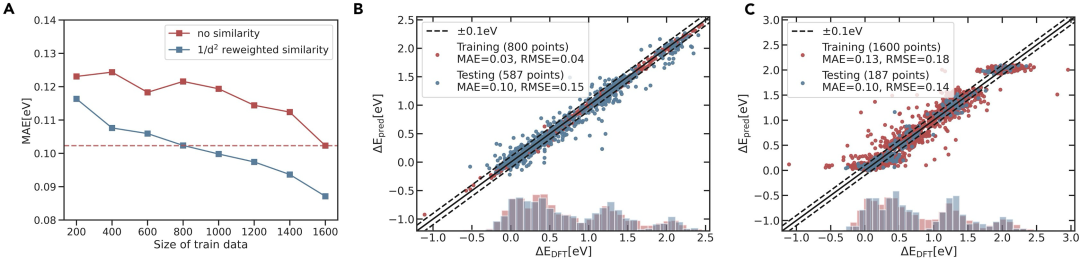
我们进一步假设可以将相似性量化作为机器学习 (ML) 训练的输入特征,以更精确地预测 OH* 和 O* 的吸附能。相比不考虑相似性的模型,三种相似性度量均提升了模型的准确性,但程度不同。改进顺序为 1/d² 加权相似性 ≥ 1/d 加权相似性 > 原始相似性,因此我们认为重新加权的相似性在结构上更具参考价值。在训练中选择 1/d² 加权相似性作为 ML 特征,将 MAE 从不含相似性特征时的 0.11 eV 降至最佳情况下的 0.08 eV。接着,我们探讨了实现特定 MAE 所需的训练集规模,并设定 0.10 eV 作为 MAE 目标。使用 1/d² 加权相似性作为最佳相似性特征进行训练,将所需的训练数据量从 1,600 个减少到 800 个,所需的全 DFT 计算量加速了 23 倍,达到了给定精度。

利用相似性信息 ML 和实验验证预测新的候选催化剂
文献信息
High-entropy alloy electrocatalysts screened using machine learning informed by quantum-inspired similarity analysis,https://doi.org/10.1016/j.matt.2024.10.001
通讯作者及单位:
Pengfei Ou, Edward H. Sargent – Department of Electrical and Computer Engineering, University of Toronto, 10 King’s College Road, Toronto, ON M5S 1A4, Canada; Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. Isaac Tamblyn - Department of Physics, University of Ottawa, 150 Louis-Pasteur Private, Ottawa, ON K1N 6N5, Canada; Department of Physics, University of Ottawa, 150 Louis-Pasteur Private, Ottawa, ON K1N 6N5, Canada.
导师简介
新加坡国立大学化学系欧鹏飞教授简介
欧鹏飞博士于2024年8月加入新加坡国立大学 (NUS) 化学系,担任校长青年教授 (Presidential Young Professor。欧博士于2020年在加拿大麦吉尔大学获得博士学位(导师:Jun Song教授),并在2020至2022年间在加拿大多伦多大学进行博士后研究,2022-2024年间在美国西北大学化学系担任Research Associate(导师:Edward H. Sargent教授)。
欧鹏飞博士长期致力于计算电化学和由数据驱动催化材料的研究和开发。作为第一作者和通讯作者(含共同)在Nat. Energy,Nat. Catal.,Nat. Commun.、J. Am. Chem. Soc.、Sci. Adv.和Proc. Natl. Acad. Sci.等学术期刊发表多篇论文。截至目前,已发表论文80余篇,被引用4000余次,H因子为35。曾获多伦多大学Climate Positive Energy Postdoctoral Fellowship和国家优秀自费留学生奖学金等荣誉。欧鹏飞博士的AI4ElectroCatalysis(AI4EC)课题组研究重点为理论指导和数据驱动的计算催化,具体研究方向包括:
催化机理研究及催化剂设计:利用第一性原理计算、过渡态搜索、微动力学建模、蒙特卡洛等理论方法,揭示与能源、环境相关的催化剂的催化机理,并实现催化剂的优化设计;
动态结构与性能关系探究:考虑电化学过程中的溶剂化、阳离子、外加电势等因素,利用分子动力学模拟和溶剂化模型对反应条件下的化学过程进行动态模拟,探究最佳的反应条件与催化剂组合;
机器学习算法和数据库开发:建立材料结构与物化性质对应的数据库,结合机器学习算法,实现纳米催化材料的高通量筛选,加速新型催化剂开发。