
引言
铜是生物体内一种重要的微量元素。铜离子可以作为辅因子参与生物体内多种酶的形成。因其具有氧化还原活性,铜可以参与细胞内的氧化还原反应,细胞呼吸和自由基的清除。在生理条件下,铜在个体中的稳态必须受到严格的调控。由ATP7A基因突变导致的个体铜缺乏会诱发门克斯疾病(Menkes disease),而由ATP7B突变导致的个体铜水平超标会诱发威尔逊氏疾病(Wilson’s disease)。在细胞水平,过量的铜会诱导细胞发生铜死亡。铜死亡是近年发现的一种新型细胞死亡方式。细胞内过量的铜离子会诱导细胞呼吸作用中的蛋白聚沉,抑制细胞呼吸,同时导致铁硫簇蛋白的减少,最终诱发细胞死亡。然而,铜离子通过何种方式进入细胞,导致铜死亡以及铜死亡在生理水平的调节意义尚不清晰。
2024年8月6日,北京生命科学研究所/清华大学生物医学交叉研究院的王晓东实验室与郑三多实验室合作,在Cell Metabolism上发表题为 Zinc transporter 1 functions in copper uptake and cuproptosis 的研究论文。该项研究首次发现并证明了Zinc transporter (ZnT1) 作为新型的铜离子转运蛋白,介导铜离子进入细胞并诱发铜死亡,并阐述了ZnT1 在肠上皮细胞中通过介导铜离子吸收,维持肠上皮Lgr5阳性干细胞稳态。该研究还为临床上用锌离子治疗铜过量引起的威尔逊氏疾病,提供了理论依据。
 在之前铜死亡的研究中,铜离子主要是通过小分子载体携带进入细胞内部。本研究中,研究人员通过使用高浓度CuSO4直接处理HeLa细胞,诱发铜死亡。在此基础上,研究人员通过CRISPR-Cas9全基因组功能缺失筛选,鉴定到锌离子转运蛋白ZnT1是铜死亡发生的必需基因。对ZnT1敲除的HeLa细胞进行CuSO4处理不能引发铜死亡,而回补表达ZnT1则可以重建铜死亡。通过ICP-MS质谱鉴定细胞内元素含量发现,相比于野生型HeLa细胞,ZnT1敲除细胞内Cu元素水平显著降低。进一步在体外脂质体实验中发现,Cu2+可以进入整合有ZnT1蛋白的脂质体内部,淬灭Calcein荧光分子,而不能进入不整合ZnT1蛋白的脂质体。这些实验证明了ZnT1具有直接转运Cu2+的功能。在后续的研究中,研究人员通过冷冻电镜结构解析与功能实验验证,揭示了Zn²⁺和Cu²⁺在ZnT1中共享主要的结合位点。Zn²⁺会竞争Cu2+与ZnT1的结合,从而有效抑制Cu²⁺转运和铜死亡。这项结果提示,临床上通过补充锌离子治疗威尔逊氏疾病,很可能是由于Zn²⁺抑制了ZnT1转运Cu2+来抑制铜吸收,从而缓解威尔逊氏疾病的症状。通过结构的比对分析,研究人员还发现,相比于其他锌离子转运蛋白家族(ZnT family)的成员,ZnT1更倾向于形成一种外向型构象(Outward-facing conformation)。这主要是由于在ZnT1二聚体中,一个单体跨膜结构域的螺旋2与另一单体跨膜结构域的螺旋3通过丝氨酸残基与谷氨酸残基形成氢键,进而稳定了外向型构象。此外ZnT1在胞外区以一种特有的亚基间二硫键,加固了这种外向型构象,更有利于铜离子的捕获和吸收。在细胞实验中,向ZnT1敲除细胞系回补上述残基的ZnT1突变体,相比于回补野生型ZnT1,均在一定程度上抑制了铜死亡,证明了ZnT1的外向型构象对于铜离子转运的功能是至关重要的。动物实验证明,肠上皮细胞中ZnT1蛋白缺失会导致小鼠锌离子吸收缺陷,从而导致小鼠死亡,但这种表型在补充锌离子后则不会发生。同时,ZnT1缺失抑制了肠上皮从血液中吸收铜,最终导致肠上皮铜含量降低,血清中铜含量升高。此外,研究人员还发现, ZnT1转运铜离子的功能对于维持Lgr5阳性肠干细胞是必需的,ZnT1的缺失会导致Lgr5阳性肠干细胞受损,而这一表型不能被外源锌离子的补充所拯救。综上所述,本研究首次发现了锌离子转运蛋白ZnT1介导铜离子转运并参与铜死亡调节。通过结构解析,提出锌离子竞争性抑制铜离子转运的分子机制,并进一步阐述了ZnT1采取一种外向型构象(Outward-facing conformation)保证了铜离子的捕获与吸收。这为临床上通过锌离子治疗威尔逊氏疾病提供了有力的理论依据。在生物体内,ZnT1转运铜离子的功能,对于肠道干细胞的维持具有重要意义。
在之前铜死亡的研究中,铜离子主要是通过小分子载体携带进入细胞内部。本研究中,研究人员通过使用高浓度CuSO4直接处理HeLa细胞,诱发铜死亡。在此基础上,研究人员通过CRISPR-Cas9全基因组功能缺失筛选,鉴定到锌离子转运蛋白ZnT1是铜死亡发生的必需基因。对ZnT1敲除的HeLa细胞进行CuSO4处理不能引发铜死亡,而回补表达ZnT1则可以重建铜死亡。通过ICP-MS质谱鉴定细胞内元素含量发现,相比于野生型HeLa细胞,ZnT1敲除细胞内Cu元素水平显著降低。进一步在体外脂质体实验中发现,Cu2+可以进入整合有ZnT1蛋白的脂质体内部,淬灭Calcein荧光分子,而不能进入不整合ZnT1蛋白的脂质体。这些实验证明了ZnT1具有直接转运Cu2+的功能。在后续的研究中,研究人员通过冷冻电镜结构解析与功能实验验证,揭示了Zn²⁺和Cu²⁺在ZnT1中共享主要的结合位点。Zn²⁺会竞争Cu2+与ZnT1的结合,从而有效抑制Cu²⁺转运和铜死亡。这项结果提示,临床上通过补充锌离子治疗威尔逊氏疾病,很可能是由于Zn²⁺抑制了ZnT1转运Cu2+来抑制铜吸收,从而缓解威尔逊氏疾病的症状。通过结构的比对分析,研究人员还发现,相比于其他锌离子转运蛋白家族(ZnT family)的成员,ZnT1更倾向于形成一种外向型构象(Outward-facing conformation)。这主要是由于在ZnT1二聚体中,一个单体跨膜结构域的螺旋2与另一单体跨膜结构域的螺旋3通过丝氨酸残基与谷氨酸残基形成氢键,进而稳定了外向型构象。此外ZnT1在胞外区以一种特有的亚基间二硫键,加固了这种外向型构象,更有利于铜离子的捕获和吸收。在细胞实验中,向ZnT1敲除细胞系回补上述残基的ZnT1突变体,相比于回补野生型ZnT1,均在一定程度上抑制了铜死亡,证明了ZnT1的外向型构象对于铜离子转运的功能是至关重要的。动物实验证明,肠上皮细胞中ZnT1蛋白缺失会导致小鼠锌离子吸收缺陷,从而导致小鼠死亡,但这种表型在补充锌离子后则不会发生。同时,ZnT1缺失抑制了肠上皮从血液中吸收铜,最终导致肠上皮铜含量降低,血清中铜含量升高。此外,研究人员还发现, ZnT1转运铜离子的功能对于维持Lgr5阳性肠干细胞是必需的,ZnT1的缺失会导致Lgr5阳性肠干细胞受损,而这一表型不能被外源锌离子的补充所拯救。综上所述,本研究首次发现了锌离子转运蛋白ZnT1介导铜离子转运并参与铜死亡调节。通过结构解析,提出锌离子竞争性抑制铜离子转运的分子机制,并进一步阐述了ZnT1采取一种外向型构象(Outward-facing conformation)保证了铜离子的捕获与吸收。这为临床上通过锌离子治疗威尔逊氏疾病提供了有力的理论依据。在生物体内,ZnT1转运铜离子的功能,对于肠道干细胞的维持具有重要意义。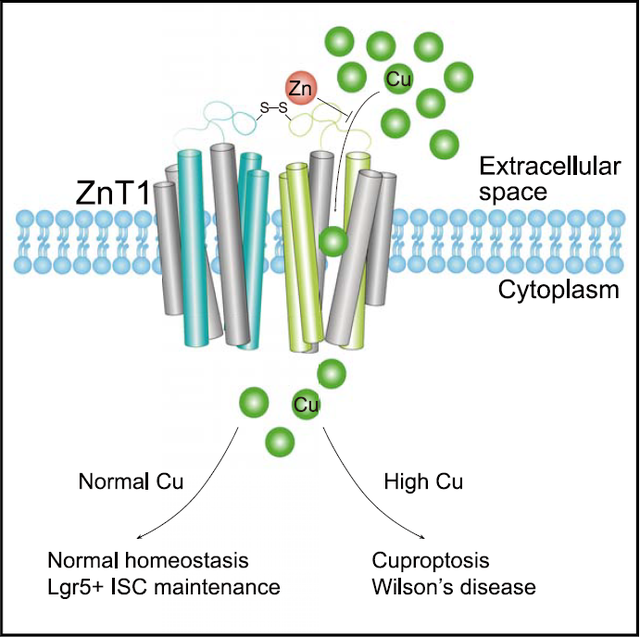 模式图(Credit: Cell Metabolism)
模式图(Credit: Cell Metabolism)参考文献
https://doi.org/10.1016/j.cmet.2024.07.009责编|探索君
排版|探索君
文章来源|“BioArt”
End