单细胞测序技术是指在单细胞水平上,通过全基因组或转录组扩增,对核酸分子进行高通量测序的技术。该技术能够揭示单个细胞的基因结构和基因表达水平,反映细胞间的异质性,剖析单个细胞对生态系统或有机体的贡献。相比传统的bulk扩增子测序和宏基因组测序,单细胞水平的微生物组测序技术能够构建单菌个体的基因组和转录组信息,研究菌个体之间的差异和相互作用。特别对于丰度相对较低的菌,也是能够获得其组学信息。目前,单细胞微生物组测序技术包括单细胞扩增子测序、单细胞基因组测序和单细胞转录组测序。其中单细胞扩增子测序(BarBIQ)能够对微生物群落进行绝对定量;单细胞微生物基因组测序(EASi-seq)可以实现对数千种微生物基因组进行一次测序;单细胞微生物转录组测序(SmRandom-seq)能够对1万个单菌细胞转录组(~1000 genes/cell)进行一次测序,实现菌种和基因表达水平鉴定。今天,小编给大家分享这几种微生物单细胞测序技术方面的“干货”,希望能给有这方面研究的老师带来一些帮助~
1.BarBIQ:基于单细菌16S rRNA的菌群高通量精准识别与绝对定量方法

摘 要
为了克服传统16S rRNA基因扩增子测序方法的局限性,金坚石团队开发了一种基于单细菌16S rRNA的菌群高通量精准识别与绝对定量方法——BarBIQ。该方法采用条形码标记单细菌的策略并实现了对16S rRNA基因鉴定的单碱基精确度。
BarBIQ与传统的16S rRNA测序方法有本质区别:首先,BarBIQ可以鉴定每个细菌的种类,并且可以定量每种细菌的细胞数量,而传统方法只能鉴定16S rRNA基因序列并量化每种基因序列的扩增数量。第二,BarBIQ可以参照测量的相对细胞数与总的细胞数的结果对菌群中每种细菌进行绝对定量,而传统方法是基于基因扩增数量的比值结合测量的总细胞数量对OTU进行绝对定量的。为使研究人员清楚了解这种基于细胞的新概念,我们引入了一个新的计量单位cOTU(cell-based OTU),即用基于细胞的16S rRNA序列类型及细胞数量来描述细菌菌群。第三,BarBIQ对16S rRNA基因的鉴定具有单碱基准确性与分辨率,而传统方法对16S rRNA基因的鉴定会出现错误。
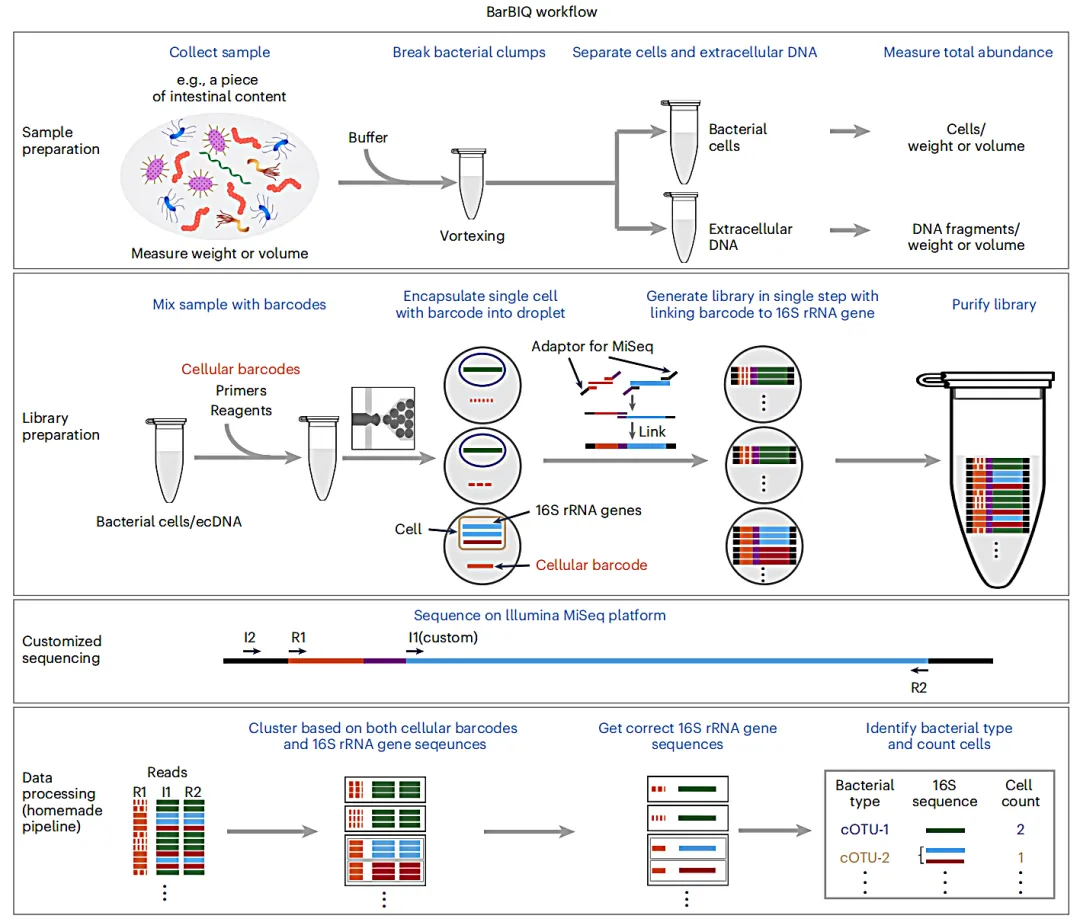
工作流程
1、将收集好的菌群样品混匀到缓冲液中,通过涡旋破碎细菌团,过滤离心分离细菌完整细胞和细胞外的DNA,利用微滴数字PCR (ddPCR)测量每毫克样本的总细胞数。
2、将细菌样本与细胞条形码、引物和试剂的溶液混合,制成液滴(直径为~120µm)进行DNA扩增。
3、随后,每个液滴中的16S rRNA基因和条形码扩增并串联连接,与此同时,将连接产物连接到测序街头接头片段。
4、回收所有液滴的文库(连接的扩增子),并通过AMPure XP beads进行两轮纯化、两轮凝胶纯化,再使用链霉亲和素包被磁珠进行两轮纯化,通过qPCR对文库进行质检和定量。
5、随后,将扩增文库与制备并纯化的spike-in文库混合后,使用MiSeq测序仪对扩增后的16S rRNA基因以及细胞条形码进行测序。
6、分析每个细胞条形码的测序结果来识别对应的菌种类型,并计算每种细菌类型的细胞数量。通过使用测量过的每毫克样品包含的细胞总数量计算出每种细菌的绝对数量(对于细胞外DNA,通过测量的每毫克的总DNA量来进行绝对定量)。
2.INVADEseq:单细胞水平上分析宿主-细菌的相互作用

摘 要
微生物组分析揭示了在主要癌症类型的人类肿瘤组织中存在微生物生态系统,包括细菌和真菌。影像学资料显示,瘤内细菌可能位于上皮细胞和免疫细胞。然而,由于细菌RNA通常缺乏poly(A)尾,因此标准的scRNAseq方法捕获肿瘤微环境中的这一微生物成分的能力有限。
为了克服这一问题,本文介绍了侵粘附定向的表达测序(INVADEseq)方法,通过引入针对细菌16S核糖体RNA基因保守区域的引物,以及真核poly(A) RNA选择的标准引物外,使用10x Genomics 5 ' scRNAseq方案。这种“附加”方法能够在真核单细胞水平上生成宿主和细菌的DNA文库,利用barcode识别单细胞和细胞内的细菌。
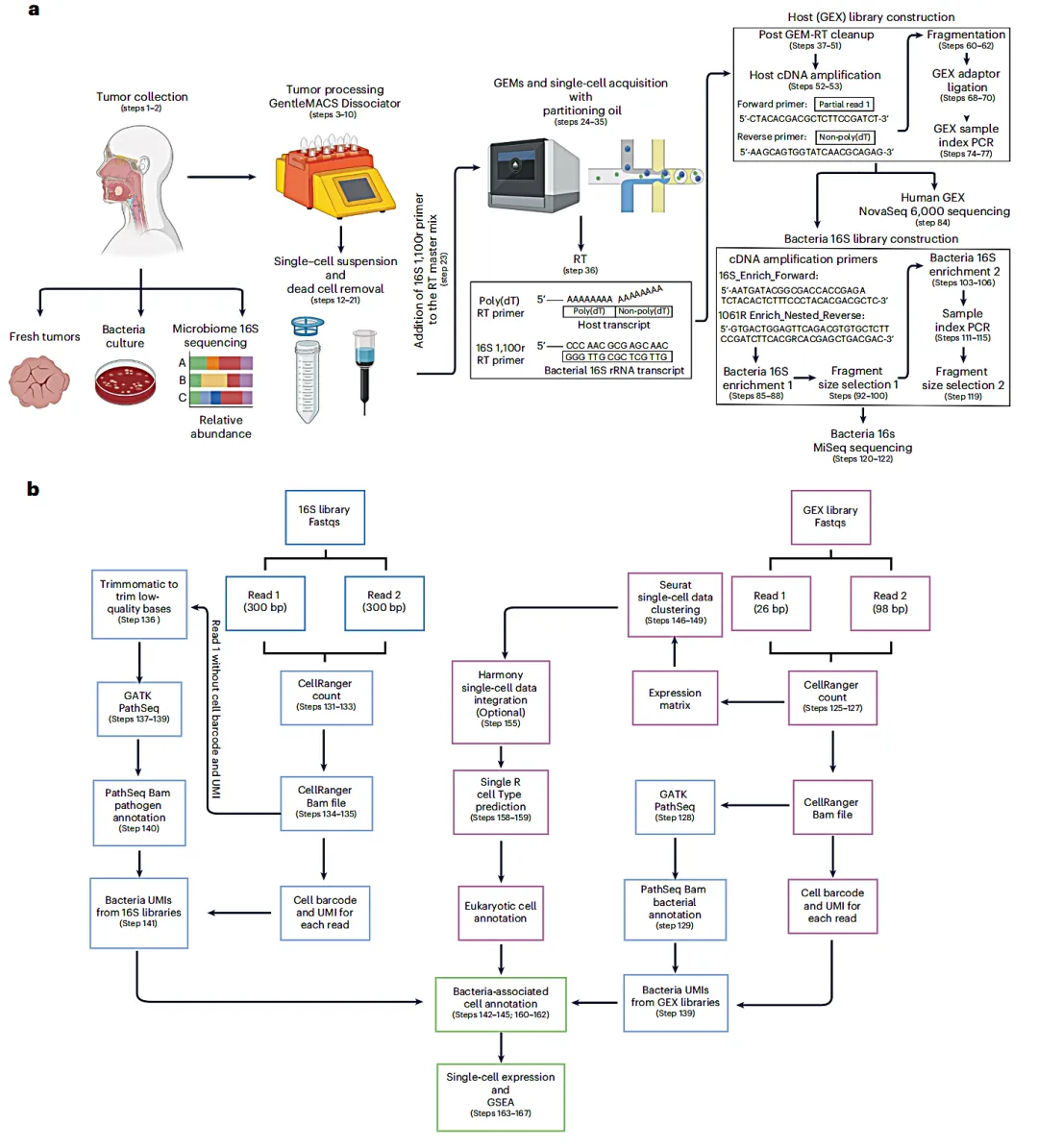
工作流程
1、采集含有微生物的肿瘤样本,通过10X单细胞5'端转录组试剂,标记单个细胞及细菌(包括黏附细胞及侵入细胞的细菌)。
2、使用含polyA引物捕获真核生物转录本,引入靶向细菌16S rRNA保守区域的引物捕获细菌转录本,然后分别构建基因表达文库GEX和16S rRNA基因文库。
3、分别针对GEX和16S数据运行cellranger,获取有效的barcode等信息,对于16S数据,进行Reads过滤后采用Pathseq软件比对,同时对配对的GEX中有效barcode信息取交集,输出交集细胞中的微生物-基因矩阵用于下游分析。
3.利用商业仪器生成的微生物组单细胞图谱

摘 要
本文中,作者介绍了EASi-seq (easy - Accessible Single microbial sequencing),一种可以高效测序上万种微生物的方法,该方法可对单个微生物的基因组进行高效测序。EASi-seq允许每次对数千种微生物进行测序,可以生成人类和环境微生物组的详细图谱。从数千个单一微生物中捕获大型基因组数据集的能力为发现和分析物种亚群提供了新的机会。为了实现这一目标,作者团队开发了配套的生物信息学流程,该流程可根据相似性将微生物聚类,改善全基因组组装、菌株鉴定、分类和基因注释。此外,该研究还展示了将宏基因组重叠群与EASi-seq数据集整合以减少捕获偏倚和增加覆盖率。总之,EASi-seq利用可在市售平台上运行的可访问的工作流程,实现了微生物组样本的高质量单细胞基因组数据。
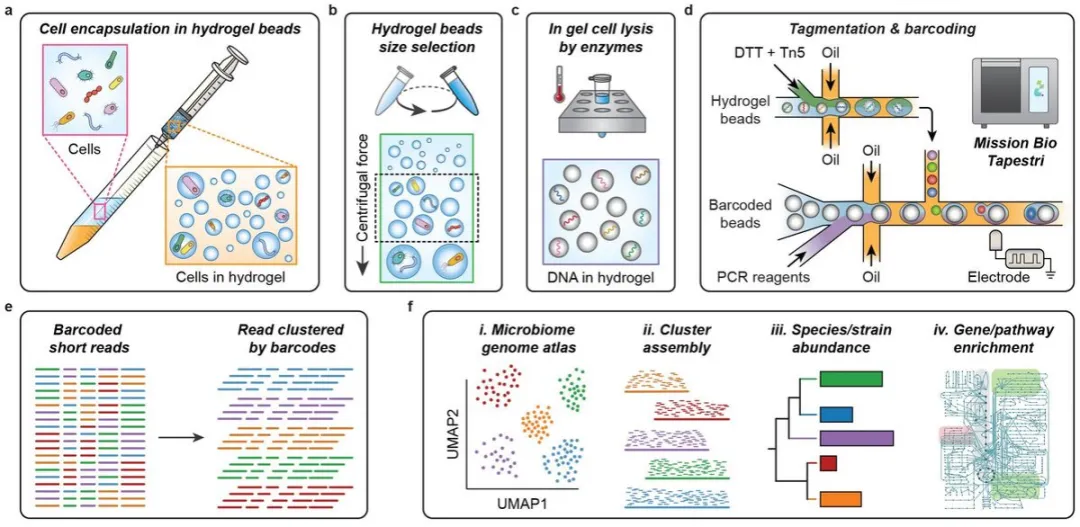
工作流程
1、Mission Bio的Tapestri是为哺乳动物细胞的靶向DNA测序而设计的,其独特之处是能够进行两个后续的液滴步骤。这项工作的一个主要创新是开发了一个微生物测序工作流程,映射到Tapestri的两步过程。
2、裂解细胞,将DNA片段分割成可读的长度,并用单细胞条形码标记片段。根据Tapestri的两步工作流程,使用第一个液滴来进行DNA标记,第二个阶段用于条形码标记。
3、使用微流控平台将单细胞封装在水凝胶球中,批量洗涤裂解细胞。利用无微流控平台在水凝胶中进行基因组纯化,通过乳化将细胞封装在水凝胶液滴中。
4、向水凝胶珠灌注由多糖消化水解酶和蛋白水解酶组成的鸡尾酒。因此,当将凝胶装载到Tapestri时,将浓度设置为每个液滴约5个凝胶,这样10%的液滴含有一个基因组,90%的液滴不含细胞,从而产生单细胞数据,并有效利用条形码液滴。
4.SmRandom-seq:基于液滴的高通量单微生物RNA-seq分析

摘 要
在本文中,作者报告了一种基于液滴的高通量单微生物RNA-seq检测方法(SmRandom-seq),该方法使用随机引物生成原位cDNA,使用液滴进行单个微生物条形码编码,并使用基于RISPR的rRNA去除进行mRNA富集。此外,该技术还成功捕获了数千个大肠杆菌个体的转录组变化,并发现了一些在抗生素胁迫下表现出不同的SOS响应和代谢通路基因表达模式的大肠杆菌耐药亚群。SmRandom-seq提供了一种高通量的单个微生物转录组分析方法,有利于促进未来在微生物耐药性、持久性、微生物-宿主相互作用和微生物组研究方面的发现。
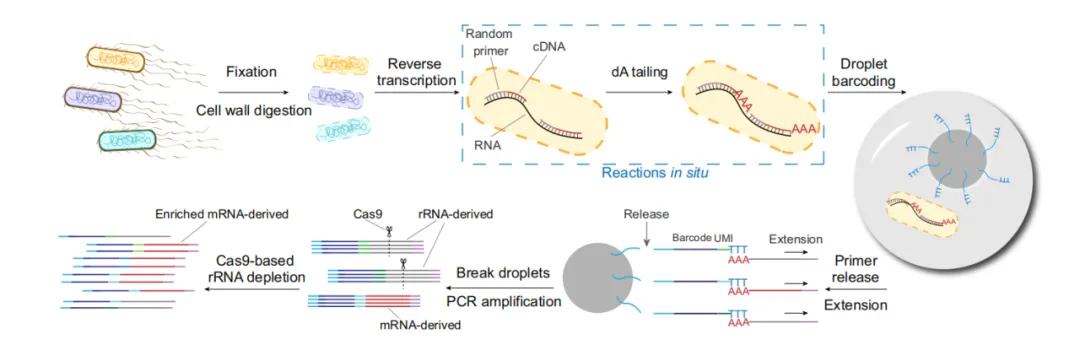
工作流程
1、首先使用4%多聚甲醛固定细菌过夜,以交联细菌内部的RNA、DNA和蛋白质。然后对固定的细菌进行透化,以便进行下一步原位逆转录反应。
2、接着加入随机引物与细菌内的RNA结合扩增总RNA。通过末端转移酶(TdT)将poly(dA)尾巴原位添加到cDNA的3’端。
3、使用微流控装置将单个细菌与标记微珠封装到液滴中,通过酶切将poly(T)引物从微珠上释放出来,同时将cDNA从细菌中释放出来。poly (T)引物与cDNA末端的poly (A)尾结合,随后延伸给cDNA加上特定标签,并给每个cDNA上添加分子标签(UMI)。破乳对标记cDNA进行扩增。
参考文献
[1] High-throughput identification and quantification of bacterial cells in the microbiota based on 16S rRNA sequencing with single-base accuracy using BarBIQ.Nat Protoc. 2023 Nov 27.
[2] NVADEseq to identify cell-adherent or invasive bacteria and the associated host transcriptome at single-cell-level resolution. Nat Protoc. 2023 Nov;18(11):3355-3389.
[3] Microbiome single cell atlases generated with a commercial instrument. Res Sq [Preprint]. 2023 Sep 14:rs.3.rs-3253785.
[4] Droplet-based high-throughput single microbe RNA sequencing by smRandom-seq. Nat Commun. 2023 Aug 23;14(1):5130.