转自:医学界
近日,复旦大学附属眼耳鼻喉科医院舒易来教授团队与韩国首尔大学医院SangsuBae、Sang-YeonLee教授团队展开国际合作。合作双方于2025年8月5日在国际期刊《自然·通讯》(NatureCommunications)上发表题为“PAM-flexibleadeninebaseeditingrescueshearinglossinahumanizedMPZL2mousemodelharboringanEastAsianfoundermutation”的研究论文。
该成果揭示了PAM灵活的腺嘌呤碱基编辑器(ABE)可挽救携带东亚创始突变的人源化MPZL2小鼠模型中的听力损失,为遗传性听力损失提供了潜在的精准治疗策略。

舒易来教授团队与合作者在NatureCommunications发表论著
耳聋是最常见的感官障碍疾病之一。据世界卫生组织(WHO)统计,全球患有致残性听力障碍的人数约4.3亿,占世界总人口的5%,其中2600万为先天性耳聋患者。新生儿耳聋的发生率约1‰-3‰,我国每年约有3万聋儿出生。
听力障碍不仅仅意味着听觉丧失,往往还伴随着言语障碍、认知发育迟缓,严重影响到患者的日常生活和社会参与。约60%的先天性耳聋是由遗传因素所导致的,目前已知的耳聋基因超过200个,然而目前暂无临床上市的治疗药物。
MPZL2突变是非综合征型常染色体隐性遗传性耳聋DFNB111(Autosomalrecessivegeneticforms111)的原因。患者主要表现出早发性、进行性的轻度至中度感音神经性听力损失,且听力损失在高频更为明显。
2018年,MPZL2首次被报道与遗传性听力损失相关。近年,一项基于大规模轻度至中度感音神经性听力损失(SNHL)儿童队列的前瞻性研究表明,MPZL2是导致遗传性轻度至中度SNHL的第二常见基因,占17.3%,仅次于STRC基因(55.8%)。值得注意的是,MPZL2中的某些特定突变(如c.220C>T)在东亚人群中具有创始效应(Foundereffect),提示其可能为该地区特有的遗传风险因素。
为了深入解析这一东亚人群中常见的遗传性耳聋突变位点致病机制,并探索更具针对性的精准治疗策略,复旦大学附属眼耳鼻喉科医院与韩国首尔大学医院联手合作。
研究团队基于临床队列研究,复旦大学附属眼耳鼻喉科医院与韩国首尔大学医院对遗传性耳聋1437例无亲缘关系的耳聋家系进行了系统性病因学分析。筛选出234例表现为对称性、轻中度非综合征型SNHL未成年患者(≤18岁),明确了155例患者的致病基因(66.2%)。
其中24例与MPZL2基因突变有关(15.5%),且23例(95.8%)患者携带至少一个c.220C>T等位基因突变。c.220C>T等位基因在东亚人群中频繁出现,提示该突变可能为东亚地区的创始突变。
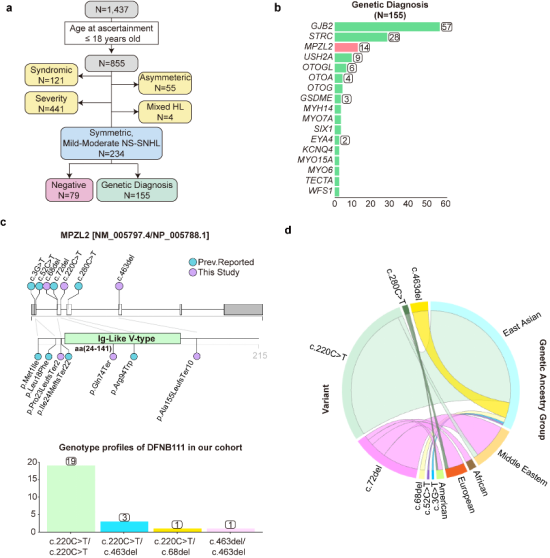
图1.东亚特异性MPZL2基因突变位点c.220C>T的确认
针对该突变位点,舒易来教授团队与合作者构建了人源化小鼠模型(hMPZL2Q74X/Q74X),该小鼠模型表现为进行性听力损失,重现了人类MPZL2耳聋病人的听力学表型。研究团队基于可实现A·T到G·C碱基编辑的腺嘌呤碱基编辑器(ABE),研发了一种具有较低旁观者编辑和脱靶效应的PAM灵活的ABE变体治疗体系:ABE8eWQSpRY:sgRNA3。
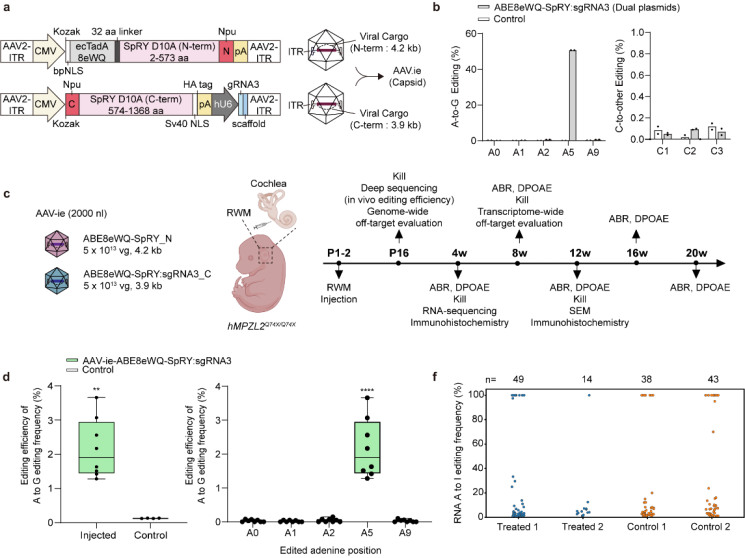
图2.安全、高效的ABE8eWQSpRY:sgRNA3治疗体系的筛选
通过双腺相关病毒(dual-AAV)的递送,研究团队成功纠正了异常基因表达,恢复突变小鼠内耳结构的完整性以及MPZL2蛋白的表达,突变小鼠的听力显著恢复并维持至少20周,且没有观察到明显的脱靶效应。该研究进一步拓宽了单碱基编辑技术在遗传性疾病中的应用范围,丰富了单碱基编辑技术的应用场景,增强了ABE技术应用于其他遗传性疾病的信心。
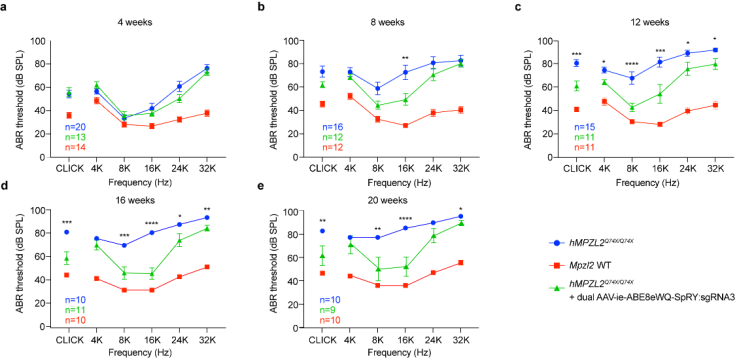
图3.ABE8eWQSpRY:sgRNA3治疗体系长期的恢复人源化MPZL2c.220C>T小鼠听力
研究团队
该研究由复旦大学附属眼耳鼻喉科医院舒易来教授团队牵头,联合韩国首尔大学医院SangsuBae团队以及Sang-YeonLee团队共同完成。
复旦大学附属眼耳鼻喉科医院胡少伟、蒋罗颖,韩国首尔大学医院的SohyangJeong以及HansolKoo为共同第一作者。
据悉,该工作是舒易来教授团队之前一项MPZL2遗传性耳聋基因治疗研究工作的延续。2024年10月,舒易来教授团队与何英姿教授团队合作在国际著名学术期刊《美国人类遗传学杂志》(TheAmericanJournalofHumanGenetics)发表题为“Hearingrestorationbygenereplacementtherapyforamultisite-expressedgeneinamousemodelofhumanDFNB111deafness”的研究论文,通过AAV-ie介导的基因替换策略,成功恢复了MPZL2基因敲除小鼠的听力及内耳结构的完整性。
舒易来教授团队与合作者在TheAmericanJournalofHumanGenetics发表论著
先天性耳聋基因治疗:开启听觉新希望
近年来,基因治疗(GeneTherapy,GT)作为一种全新的、针对病因的治疗手段,有望恢复自然听力,引起了领域内的高度关注。基因治疗,简单来说,就是通过生物技术手段,将正常基因或者修复异常基因的工具导入患者细胞内,替代或修复那些导致疾病的缺陷基因,从而从根源上解决问题。
复旦大学附属眼耳鼻喉科医院耳聋基因治疗团队长期深耕此领域,创新性采用双AAV载体递送系统,采用基因置换策略,将正常的人源OTOF编码序列导入患者内耳感受声音的毛细胞,促使毛细胞能够表达正常功能的耳畸蛋白,从而从根本上改善听力和言语。
经过数年刻苦攻关,成功研发出OTOF耳聋基因治疗药物,2022年6月,临床试验获伦理委员会批准;同年,成功完成全球首例耳聋患者的基因治疗药物体内给药。此后,又陆续为10余名患者给药,成功纠正患者听力和言语,这是国际首个先天性耳聋基因治疗临床试验(First-in-Human),也首次实现了双AAV载体人体递送,解决了大基因递送这一医学难题,开启了耳聋基因治疗新时代,为全球耳聋患者带来“听见世界”的曙光。
基因治疗背后的探索之路
这些成果的取得,离不开团队长期不懈的努力。创新性运用基因置换、基因编辑等前沿技术,在多个耳聋模型上证明了基因治疗的疗效。
此前应用于临床突破的针对OTOF耳聋基因突变所致的耳聋,攻克了两大挑战。
首先是递送系统的限制。AAV是最常用的基因治疗递送载体,但OTOF基因编码序列超出了单个载体的装载容量。研究团队采用双AAV载体拆分递送策略,成功突破单载体基因容量限制的技术瓶颈,在耳蜗细胞内实现基因序列的精准重组。
二是给药困难。耳蜗位置深、细胞种类多,且被骨质结构包绕。针对耳蜗靶向递送的技术挑战,团队经过反复摸索,自主研发出了一套精准、微创的内耳递送路径和给药装置,为耳聋基因治疗提供了重要技术保障。
经过多年探索和研发,团队率先开展了全球首个先天性耳聋基因治疗的临床试验,成功纠正了聋哑患儿听力、言语和声源定位能力,相关研究结果已发表于顶级医学期刊《柳叶刀》和《自然·医学》等杂志,获得《柳叶刀》的同期述评,并入选封面导读,国际同行评论道“为耳聋治疗提供了范式转变,并为治疗其他形式的听力损失带来希望”“标志着基因治疗听觉障碍乃至更广泛疾病的新时代开启”。
舒易来医生在国际著名杂志《自然综述遗传学》(NatureReviewsGenetics)发表题为:Genetherapyfordeafness:wecandomore(耳聋基因治疗:我们可以做更多)的评论文章,就目前耳聋基因治疗的发展现状以及未来挑战与展望进行了专题论述。
此外,舒易来教授团队研究发现,基因治疗后受试者听觉皮层在音乐和言语刺激时激活显著提升,特别是在前颞叶、颞顶叶区域,提示听觉通路功能重建,听觉皮层对不同类型声音的处理功能逐渐恢复;而且,基因治疗恢复耳畸蛋白缺陷患者自然听力,在噪声言语与音乐感知中比人工耳蜗表现更优。这些研究成果发表于国际权威期刊《自然·人类行为》(NatureHumanBehaviour)以及《美国医学会·神经病学》(JAMANeurology)等杂志。
团队成员在基因治疗后合影