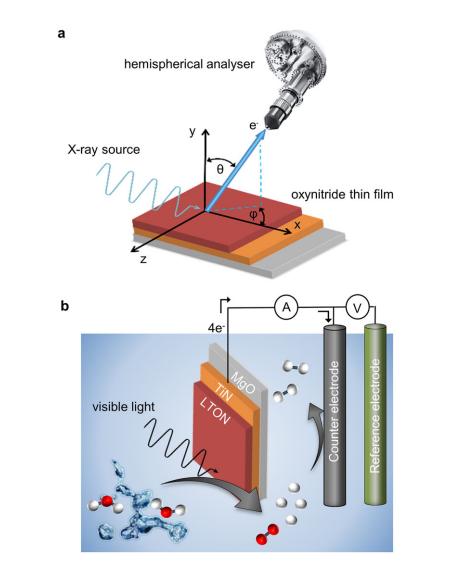
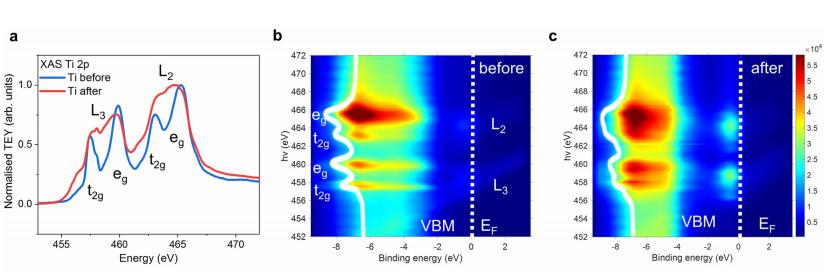
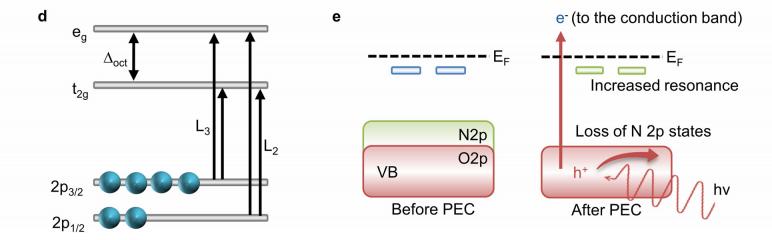
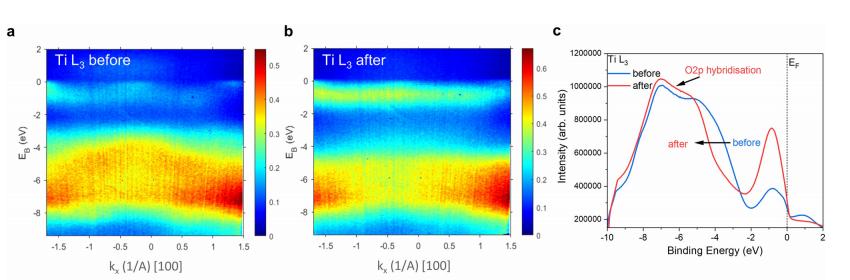
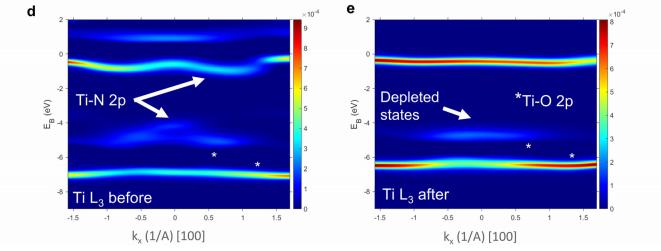
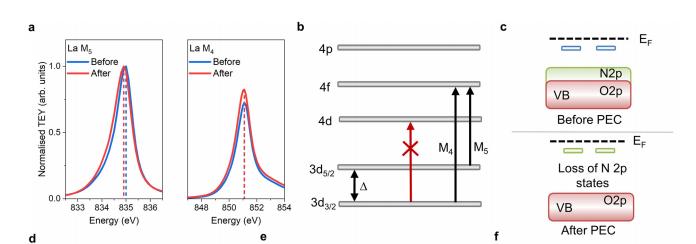
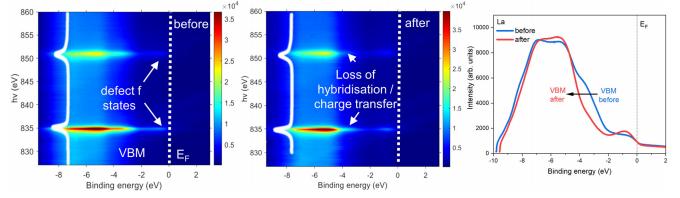
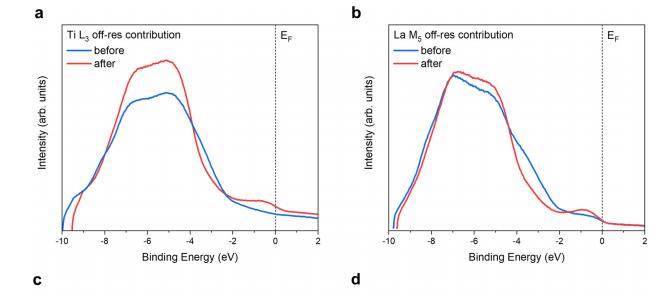
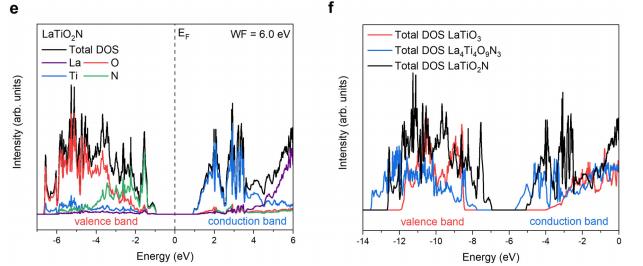
共振软x射线ARPES对LaTiO2N光催化剂动量分辨电子结构的稳定性 前言:近年来,可见光驱动的水分解作为一种绿色能源生成方法备受关注,其中氧氮化物被认为是潜在的高效光催化材料。 在这些材料中,氧氮化钛镧(LaTiO2N)因其优越的光催化性能和稳定性而备受瞩目,然而,要深入理解这些材料的光催化机制以及电子结构的变化却面临着一些挑战,尤其是在光电化学反应条件下。 我们经过DFT计算经由实验的ResPE数据证实了以下结果:在费米能级EF(0 eV)以下,价带最大值(VBM)(-3到-4 eV)由与N 2p态杂化的Ti和La态组成,而在价带底部,这些态与O 2p态杂化。 在这里更容易跟踪LTON中DOS的演变以及与OER相关的变化,我们发现与ResPE图中的观察结果相一致,并观察到PECR后总体谐振强度增加,这表明在VB底部(约-5到-8 eV)中产生了额外的O 2p态或更大程度的杂化。 我们还观察到VBM(-3到-4 eV)的强度降低,这是因为在OER条件下,在LTON的表面附近/附近部位丢失了部分晶格中的氮,这意味着由于丧失了靠近VBM附近的态,光子在可见光区域的吸收概率降低。 我们先是比较了Ti(c)和La(d)的谐振贡献,表示LTNO的价带中Ti和La杂化态的权重,发现了光电化学反应(PECR)前后。 在PECR后,我们可以看到VBM(-3 eV)处La和Ti权重的轻微损失,我们将这些谱的演变归因于在OER期间表面丢失晶格中的部分氮,导致VBM(-3 eV)附近的La-N和Ti-N杂化态部分耗尽。 我们为了与PECR后的LTNO的演变进行比较,我们将化学计量比的LTON的计算总DOS与不稳定的LaTiO3(LTO)和阴离子缺陷结构La4Ti4O9N3(D-LTON)进行了比较。 为了清晰起见,本比较中仅包含了自旋向上的贡献,这些材料的选择与它们各自的元素组成相对应,以反映出在失去氮并产生阴离子空位和/或改变氧/氮含量的情况下,LTNO的电子结构可能的趋势,正如谐振SX-ARPES数据在PECR期间的演变所示。 我们为了更准确地比较这三种材料,它们的DOS的能量尺度被对齐到费米能级EF(0 eV),我们注意到LTO和D-LTON的DOS显示这些材料的EF(0 eV)位于CB中,由反键态组成。 它们的占据是不利的,因为它会破坏晶格,导致其重新组织,通常这种重新组织通过相变或由于空位形成进行,这解释了为什么LTO不以自然矿物的形式存在。 我们还注意到LTO和D-LTON都是自旋极化的,因为LTO中的Ti的形式电荷为3+,而D-LTON是一个非化学计量比的材料。 我们可以看到阴离子缺陷的D-LTON或不稳定的LTO中N的部分或全部丧失将VBM能量向下移动(约2和4.5 eV),这进一步表明,在OER期间,O/OH-中间体参与了从表面晶格N原子产生硝酸盐和亚硝酸盐物种的光催化形成,它们离开晶格并以NOx物种的形式在表面上以化学吸附形式存在。 这种效应与CB中电子色散在费米能级附近的抑制是一致的,我们推测在OER期间,由于从晶格结构中失去N而生成的剩余氧空位愈合,O原子可能会扩散到LTNO中,由O*和OH-反应中间体生成,这与PECR后的非共振谱中VB强度(约-4到-7 eV)略微增加一致。 然而,Ti和La的谐振加权贡献显示在价带底部La的谐振强度增加,表明La-O 2p杂化增加,而Ti的谐振强度减少。 这可以通过许多这样的氧化物和氮化氧化物半导体的表面优选AO末端(在LTNO的情况下为LaO)来解释,这是通过低能离子谱学实验确定的。 通常情况下,作为氧气进化的活性位点的B阳离子是过渡金属阳离子,会被隐藏在第一个原子表面层之下,因此,表面的氧氮空位会暴露出底层的过渡金属阳离子。 先前的计算研究表明,对于LaTiO2N53,更多的氧气加入La-O表面层,SC就会自掺杂电子(累积),填充空的Ti 3d态。 我们在La的谐振光谱中看到了这种效应的某些证据,显示增加的O杂化,而Ti的谐振光谱则显示PECR后在费米能级(0 eV)以下的Ti 3d权重的增加。同样的工作53还提出,表面的演化会导致一种几何配置,其中两个N原子离开Ti层并移到La表面层,导致更稳定的5配位Ti构型。 结论:先前的工作表明,LTNO会发生表面重构,Ti显示出产生空位的迹象(导致欠配位的Ti),但其周围的电荷被保持不变。 共振软X射线ARPES技术为研究LaTiO2N光催化剂的电子结构提供了有力的工具,我们通过对材料的能带结构、能隙以及表面态等性质的探究,能够更好地理解其光催化性能,并为光催化材料的设计和应用提供重要的指导,这项研究为开发高效的可见光催化剂以解决能源和环境问题提供了有价值的信息。