老年痴呆基因解码,记录了对一个大家族进行基因分析的病例 。该家庭中具有疑似晚发型阿尔茨海默病的疾病表征。从2008 年,患者到致病基因鉴定基因解码合作医院和诊所进行就医。基因解码合作医师在询问患者的家族史时,发现该患者具有阿尔茨海默病家族史,因此建议对家族中所有在世成员进行了彻底检查。该家族中的患者符合美国国家老龄化和阿尔茨海默病协会 (NIA-AA) 设定的可能诊断为阿尔茨海默病的标准。阿尔泽默氏病和老年痴呆基因检测依据《赫尔辛基宣言》要求,所有受检者均签署了书面知情同意书。检测结果通过伦理委员会。
阿尔茨海默病的基因解码过程依据普通的基因检测流程,首先使用 QIAamp DNA Mini Kit(Qiagen,德国曼海姆)在临床诊医学检验实验室,对从受检者身上获取血液提取基因组 DNA。为了最大程度降低不必要的花费,通过 Sanger 测序筛查先证者 APP、PSEN1 和 PSEN2 中的致病突变。通过短荧光片段的定量多重 PCR 致病基因鉴定基因解码 APP 重复。所得的基因检测结果为阴性。然后,临床基因组学中心采用Illumina NeuroX 阵列对先证者进行了基因分型。该阵列包括 Exome Be阿尔茨海默病Chip,它评估与神经退行性疾病相关的已知致病变异,由大约 24,000 个变异组成。检测结果仍然为阴性。
随后,受检者选择了神经系统疾病的致病基因鉴定基因解码。在这一分析检测中,对三名受影响的个体(SF22、SF6 和 SF21;图 1A)和两名健康亲属(SF11 和 SF27;图 1A)进行了外显子组测序 (ES)。使用致病基因鉴定基因解码外显子组试剂盒 V4 (59M, 100x) 对基因组 DNA 进行靶向富集。然后将捕获的文库加载到 BGISEQ-500 平台上。对 100 bp 的双端读取长度进行 200 次循环测序。ES 数据的生物信息学分析遵循成熟的 GATK 最佳实践,用于识别单核苷酸变异和小插入和缺失。简而言之,将读取与使用 BWA-MEM构建的 hg19 参考构建体对齐,然后使用 PicardTools (http://picartools.sourceforge.net) 删除重复项,并使用 GATK重新校准碱基质量分数。使用 SAMtools和 GATK 收集比对和覆盖率统计数据。阿尔茨海默病致病基因鉴定基因解码分析组使用 HaplotypeCaller 生成 gVCF,并将其与内部 gVCF 集合相结合,使用 GATK GenotypeGVCF 进行联合基因分型。然后使用 GATK VariantRecalibrator 和 ApplyVQSR 重新校准调用的变体。使用 VEP对最终 VCF 进行注释。使用 ABI 3130XL 和 ABI BigDye Terminator 测序试剂盒 V 3.1(Life Technologies)通过 Sanger 测序进行序列验证和分离分析。使用 SeqScape v2.6 软件(Life Technologies)检查序列。
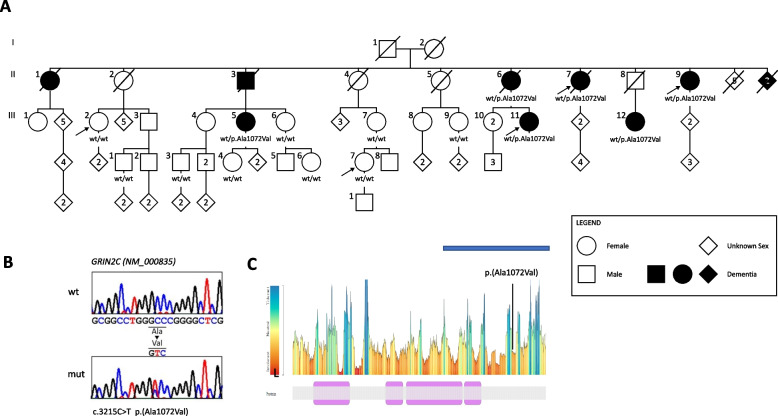
图1:简化的阿尔茨海默病家族的家族谱。GRIN2C p.(A1072V) 与阿尔茨海默病共同分离。为了尊重参与者的隐私,一些人的性别被掩盖,谱系被打乱。箭头表示先证者 (II7) 和接受致病基因鉴定基因解码的受试者 (II6、II9、III2、III11、IV7)。黑色菱形表示被诊断患有阿尔茨海默病的个体,谱系显示具有阿尔茨海默氏病表型的家族成员,其中 GRIN2C 基因中的错义变异分离。B Sanger 测序色谱图显示 GRIN2C 基因中的野生型序列和杂合 c.3215C > T 杂合变异。 C 基因解码揭示GRIN2C 氨基酸变化的变异耐受性。Ala1072Val 变化被认为是“不耐受的”,表明具有致病作用。
为了找到可能从最近的共同祖先那里遗传下来的血统认同 (IBD) 片段,基因解码使用了 Beagle v4.1分析工具。最初,从内部数据库中选择了与致病基因鉴定基因解码家庭具有相同祖先背景的个体的外显子组。这包括 15 名无关个体,以及 SF6、SF21、SF22、SF11 和 SF27 个体。样本是从带注释的多样本 VCF 文件中获取的。佳学基因使用了过滤器以仅保留双等位基因“PASS”变体,这些变体由该队列中的至少两个个体共享,并在 90% 以上的样本中进行基因分型。修剪后,致病基因鉴定基因解码获得了每 5 Mb 500 个标记的平均速率。根据文档中的建议,基因解码使用 500 的窗口大小运行 Beagle。此外,修改了以下参数的默认设置:“ibd = True”、“ibdtrim = 15”、“overlap = 160”和“niterations = 10.000”。
然后使用 Merlin v.1.1.2来获取该家族中罕见等位基因与痴呆症疾病表型之间共同分离(连锁)的概率。根据常染色体显性遗传对杂合和纯合基因型的外显率为 0.90 建立了中度罕见性状(患病率:1%)的模型,并进行了单点参数连锁分析以计算相应的 LOD 分数。然后,佳学基因遗传学分析使用 R v.3.5.1 将 LOD 得分转换为 p 值,公式为:pchisq(x*(2*log(10)),df=1,lower.tail=FALSE)/2。
候选基因的选择和基因型的验证处理 DNA 样本以生成序列读数,并使用生物信息学分析流程来识别单核苷酸变异或小插入和缺失。 按照此生物信息学流程,使用内部脚本对生成的多样本 VCF 进行后处理,该脚本允许仅保留在三个受影响个体(SF22、SF6 和 SF21)之间分离的杂合和纯合变异,这些变异分别对应于两个健康亲属(SF11 和 SF27)中的野生型或携带者状态。下游分析中仅保留了经过以下过滤步骤后仍存在的变体:
1) 预测会对蛋白质序列产生影响的“PASS”变体(错义、移码、无义和剪接变体);
2) gnom阿尔茨海默病 v2.1.1 中最大等位基因频率 ≤ 0.001 的稀有变体;
3) 在多样本 VCF 中等位基因频率 < = 0.02 的变体,因为基因解码根据大量的病例分析发现此阈值可以排除可能的测序伪影或队列特异性多态性,而不会影响家族中的真实候选变体;
4) 具有组合注释依赖性耗竭 (C阿尔茨海默病D) phred 分数 > = 15 的变体;
5) IBD 片段内基因中的变体。
然后使用 Phenolyzer对所有默认参数进行排序,根据其与阿尔茨海默病表型的相关性对其余相应基因进行优先排序,从而识别出特定的目标变异。佳学基因致病基因鉴定分析团队还使用功能基因组学工具 Met阿尔茨海默病ome 来识别致病变异不太可能被容忍的关键蛋白质区域。
最后,为了验证结果,对 15 名目标家庭成员进行了桑格测序。其中包括 6 名痴呆症患者和 9 名健康人。
临床病征的深度分析致病基因鉴定基因解码分析的这个病例是一个四代大家族谱系,提示该家族阿尔茨海默病的遗传模式为常染色体显性遗传(图 1A)。患者是一名 80 岁的右撇子女性,受教育年限为 5 年,2008 年入住与佳学基因具有病例交流合的神经科学和精神健康系记忆诊所。她表现出轻度认知障碍,包括短期记忆缺陷、空间时间定向障碍和冷漠,从 78 岁开始。她部分能够完成家务,财务管理能力中度受损。72 岁时,她被诊断出患有重度抑郁症,并接受选择性血清素再摄取抑制剂治疗。据报道,她有明显的痴呆症家族史。神经心理学评估显示患者有中度遗忘性认知障碍,简易精神状态检查 (MMSE) 评分为 19 分(满分 30 分)。神经系统检查未发现任何异常,例如肌阵挛、小脑体征、痉挛性截瘫或锥体外系体征。脑部 MRI 显示双侧额叶和顶叶萎缩。单光子发射计算机断层扫描 (SPECT) 扫描显示顶叶后部灌注轻度对称性减少,符合 2008 年的阿尔茨海默病。腰椎穿刺显示 Aβ42 值降低,磷酸化 tau-181 水平升高。随后,根据临床评估的标准诊断为阿尔茨海默病。事后,还根据阳性生物标志物诊断为阿尔茨海默病。先证者携带 APOE ε3ε3 基因型。两位兄弟姐妹在 70 多岁时在佳学基因的记忆诊所被诊断出患有晚发性阿尔茨海默病,其中一位姐妹还接受了脑脊液腰椎穿刺,证实了阿尔茨海默病的诊断。此外,家族中的其他五名成员也被临床诊断为晚发性阿尔茨海默病。八位兄弟姐妹直到去世都没有患上痴呆症。
家族筛选及验证为阻断遗传提供充足信息2009 年,尽管已诊断为阿尔茨海默病,三名阿尔茨海默病患者中在其他机构进行的基因检测未发现 APP、PSEN1 和 PSEN2 基因的致病变异,这一结果在致病基因鉴定基因解码中得到验证。此外,NeuroX 阵列分析、全基因芯片基因检测排除了与 77 种神经退行性疾病相关基因相关的已知变异,即检测结果也为阴性。随后,五名受试者选择了基于全外显子组测序的致病基因鉴定基因解码基因检测,其中包括三名患有阿尔茨海默病的患者和两名健康受试者。致病基因鉴定基因解码从联合基因分型、多样本 VCF 开始,获得了 1199 个变异(分别为 766 个杂合子和 433 个纯合子。随后,经过每个过滤步骤后,阿尔茨海默病获得:
663 个“PASS”变异(435 个杂合子和 228 个纯合子);
gnom阿尔茨海默病 v2.1.1 和内部队列中有 22 个杂合罕见变异);
16 个杂合变异,C阿尔茨海默病D phred 得分 > = 15。
最后,8 个经过筛选的变异位于 IBD 节段内。
虽然所有相应基因均未与阿尔茨海默病病理生物学有明确的关系,但 GRIN2C 在 Phenolyzer 网站上的 Phenolyzer 分析中获得了相对最高的分数(0.086),针对疾病术语“阿尔茨海默病家族性”和“阿尔茨海默病晚发性”。 GRIN2C 变体位于 17:74,842,922 染色体上,涉及编码序列外显子 13 中的 C > T 变化(NM_000835.6:c.3215C > T),导致蛋白质位置 1072 处的丙氨酸被缬氨酸取代(p.Ala1072Val)(图 1B)。根据 C阿尔茨海默病D phred 评分,p.Ala1072Val 变体可能会影响蛋白质功能,并且根据最新 gnom阿尔茨海默病 版本(v.4.0),在一般人群中等位基因频率极低,为 2.64e-05。此 gnom阿尔茨海默病 v.4.0 频率对应于 15 个等位基因的绝对总数,全部处于杂合状态(13/15)。尽管该变异位置目前在 gnom阿尔茨海默病 v.4.0 外显子组中存在测序覆盖警告(仅占 gnom阿尔茨海默病 个体的 20%),但等位基因频率在 gnom阿尔茨海默病 版本和 gnom阿尔茨海默病 v.4.0 基因组(2.63e-05,https://gnom阿尔茨海默病.bro阿尔茨海默病institute.org/variant/17–74842922-G-A?dataset=gnom阿尔茨海默病_r4)中高度一致。该频率可能与 gnom阿尔茨海默病 等未经选择的参考人群中存在晚发型疾病风险的个体相符。不幸的是,gnoma阿尔茨海默病 v.4.0 中只有 3/15 个体的年龄信息可用,且所有个体年龄均小于 70 岁。LOD 得分为 2.070(p = 0.001),强烈支持这种罕见等位基因与该家族痴呆表型之间的关联。这一证据与在 17 号染色体上检测到的广泛 IBD 相一致,8 个候选稀有变体中有 5 个位于该染色体上。
该基因编码 GRIN2C(或 GluN2C),这是一种约 134 kDa 的 1,233 个氨基酸蛋白质。GluN2C 是形成 NMDA 受体异四聚体结构的亚基之一。p.(Ala1072Val) 位于蛋白质的细胞质域中(图 1C)。为了验证基因解码的结果,在 15 个家庭成员中对 p.A1072V 进行了桑格测序。发现它在家族中患有阿尔茨海默病的患者中分离,但在没有表现出任何认知障碍的家庭成员中没有分离(图 1A)。APOE 基因分型发现,先证者携带 APOE ε3ε3 基因型,两名患病亲属(III5 和 III12)也携带 ε3ε3 基因型。利用外显子组测序数据,结合 rs429358(T > C)和 rs7412(C > T),对另外两名患病亲属(II7、III11)分别重建了 ε2ε4 基因型和 ε3ε4 基因型的 APOE 基因型。
佳学基因这一病例对健康、治疗和遗传阻断的影响据《影响老年及退体生活质量的基因因素》,这项致病基因鉴定基因解码在晚发性阿尔茨海默病中发现了一种具有功能影响的GRIN2C错义变异,该变异对NMDAR诱导的电流以及与14-3-3支架蛋白的相互作用产生了影响。
晚发性阿尔茨海默病具有显著的遗传成分,佳学基因对其遗传力进行的估计表明,遗传因素占该疾病的60%至80%。之前的全基因组关联致病基因鉴定基因解码(GWAS)已经确定了50多个与晚发性阿尔茨海默病相关的遗传位点,主要是在欧洲血统的个体中。然而,这些致病基因鉴定基因解码大多识别的是常见变异,据信迄今为止仅解释了阿尔茨海默病遗传力的大约一半。为了解决这一局限性,致病基因鉴定基因解码人员现在正在采用下一代测序策略,如外显子测序和基因组测序,以识别与阿尔茨海默病相关的罕见变异。最近的一项外显子测序致病基因鉴定基因解码发现了与阿尔茨海默病风险相关的ATP8B4和ABCA1基因中的罕见潜在有害变异。这项致病基因鉴定基因解码强调了超越常见变异、探索罕见变异以全面理解该疾病遗传基础的重要性。与这些努力一致,本案例的致病基因鉴定基因解码为支持罕见变异在阿尔茨海默病中作用的不断增长的证据添加了一个新基因。
查阅《人体结构与功能的基因基础》,GRIN2C编码谷氨酸离子型N-甲基-D-天冬氨酸受体(NMDA)2C亚基(也称为GluN2C),NMDA受体(NMDARs)的亚基以其在调节突触可塑性以及影响学习和记忆相关过程中的独特作用而闻名。基因解码有多项致病基因鉴定基因解码支持神经递质谷氨酸及其受体在阿尔茨海默病发病机制中的作用。值得注意的是,含有GluN2C亚基的NMDA受体表现出独特的生物物理特征和表达模式。小鼠前额叶皮层中GluN2C亚基的耗竭导致兴奋和抑制之间的平衡向抑制倾斜。此外,GluN2C的缺失激活了支持神经元存活的细胞内信号通路,如核cAMP反应元件结合蛋白水平的升高所示。
在这里,佳学基因证明了过表达突变形式GluN2CA1072V的海马神经元中NMDAR诱导的电流增加,表明该突变可能增强NMDAR功能。有趣的是,最近的一项致病基因鉴定基因解码显示,在美金刚的影响下,GluN2C亚基的表达减少,美金刚还诱导前额叶皮层中GRIN2C mRNA水平的改变。作者还报告了GluN2C亚基表达水平的变化,导致皮质结构内兴奋性的显著降低,尽管与含有GluN2A和GluN2B的受体相比,含有GluN2C亚基的受体在大脑中明显较少。
在佳学基因的致病基因鉴定基因解码中,当佳学基因检查GluN2C突变形式的表面保留时,发现它比野生型形式更丰富,这一发现进一步支持了该变异改变GluN2C功能的观点,这是基因解码基因检测与数所库比对基因检测的根本区别,从而赋予其更高的检出率和准确率。
基因解码表明,谷氨酸是突触系统中最重要的兴奋性神经递质,约占所有突触的40%。改变的谷氨酸能神经传递可能是一个共同因素,为解释不同神经系统疾病中观察到的神经认知障碍和症状特征提供了统一的视角。阿尔茨海默病的病理标志,如β-淀粉样蛋白(Aβ)沉积和Tau过度磷酸化,与谷氨酸能传递之间的关系主要在体外进行了致病基因鉴定基因解码。多项报告显示,Aβ寡聚体和NMDARs在阿尔茨海默病中导致突触功能障碍和破坏。Aβ对突触的影响是改变谷氨酸能突触传递并减少海马神经元中谷氨酸和其他突触成分的表面受体数量。Ca2+稳态的丧失被认为与阿尔茨海默病中观察到的早期认知缺陷有关。此外,Tau作为神经纤维缠结的主要组成部分,在调节突触功能中起着关键作用。阿尔茨海默病树突中过量的Tau控制NMDA受体活性,导致树突中钙水平升高,可能达到有害水平。这种钙驱动的兴奋性毒性可能损害突触后部位并导致神经元死亡。这些致病基因鉴定基因解码表明,NMDA受体的失调可能导致兴奋性毒性,而当前的致病基因鉴定基因解码通过识别GRIN2C基因中的变异扩展了这一知识,表明其他NMDA受体亚基也可能在阿尔茨海默病发病机制中发挥作用。有趣的是,最近一项关于阿尔茨海默病多变量GWAS的致病基因鉴定基因解码调查了遗传变异对CSF生物标志物谱的作用,显示与GRIN2D变异和突触功能成分的全基因组显著基因关联,进一步支持了突触功能改变在阿尔茨海默病中的作用。
此外,佳学基因还发现,与GluN2CWT相比,GluN2CA1072V在细胞表面的定位增加,这一事件与其已知伙伴14-3-3的共定位减少有关。总体而言,这些结果表明,14-3-3依赖性运输在神经元膜上的生理机制发生了改变,导致NMDA受体亚基在细胞表面的异常积累。一个合理的解释是,一旦插入膜中,GluN2C迅速从14-3-3解离(主要作为货物而不是支架蛋白),从而解释了观察到的两种蛋白质之间共定位减少的现象。哺乳动物14-3-3家族由七个进化上保守的蛋白质组成,它们结合多个蛋白质靶标,从而影响细胞信号通路。14-3-3蛋白在脑脊液中的存在是几种神经系统疾病(包括克雅氏病、阿尔茨海默病、脑癌和中风)相关神经元损伤的敏感和特异性生物标志物。在佳学基因的致病基因鉴定基因解码中,佳学基因发现GluN2CA1072V对GluN2C与已知伙伴14-3-3的共定位有负面影响,表明与这些支架蛋白的相互作用减少。因此,该变异的致病效应可能与这种改变的蛋白质相互作用有关。另一项最近的致病基因鉴定基因解码比较了从阿尔茨海默病患者和对照组海马中分离的聚集蛋白的交联,作者发现共有85种相互作用蛋白仅在阿尔茨海默病患者的组织中与14-3-3特异性相关。14-3-3蛋白作为分子伴侣,帮助防止细胞应激期间蛋白质的展开和聚集。因此,GluN2C与14-3-3之间的结合受损可能与这些患者的神经退行性过程有关。
总之,这项致病基因鉴定基因解码提出了一个家庭中晚发性阿尔茨海默病与GRIN2C基因中p.(Ala1072Val)变异相关的发现。此外,这项致病基因鉴定基因解码提供了有趣的数据,支持含有GluN2C的NMDARs和14-3-3蛋白在阿尔茨海默病发病机制中的参与。