
1 Nat. Commun.:单原子距离效应对钴氧化物酸性OER催化剂稳定性的影响

开发高效经济的酸性析氧反应电催化剂是质子交换膜水电解槽(PEMWE)发展的关键。钴氧化物因其高活性而被认为是很有前途的非贵OER催化剂。然而,Co原子在酸性介质中的严重溶解会导致晶体结构的失稳,这阻碍了它们在PEMWE中的应用。 中国科学技术大学曾杰、周仕明等人报道了在尖晶石钴氧化物晶格中引入耐酸Ir单原子可以显著抑制Co的溶解,并使其在酸性OER过程中保持高度稳定。结合理论和实验研究,发现Ir杂原子诱导的稳定效应与相邻Ir单原子的距离有很强的依赖性,随着距离的减小,钴氧化物的OER稳定性不断提高。当距离减小到0.6 nm左右时,尖晶石钴氧化物在酸性OER中60小时的稳定性测试中没有出现明显的降解,具有实际应用的潜力。 相关工作以《Distance effect of single atoms on stability of cobalt oxide catalysts for acidic oxygen evolution》为题在《Nature Communications》上发表论文。
图文导读
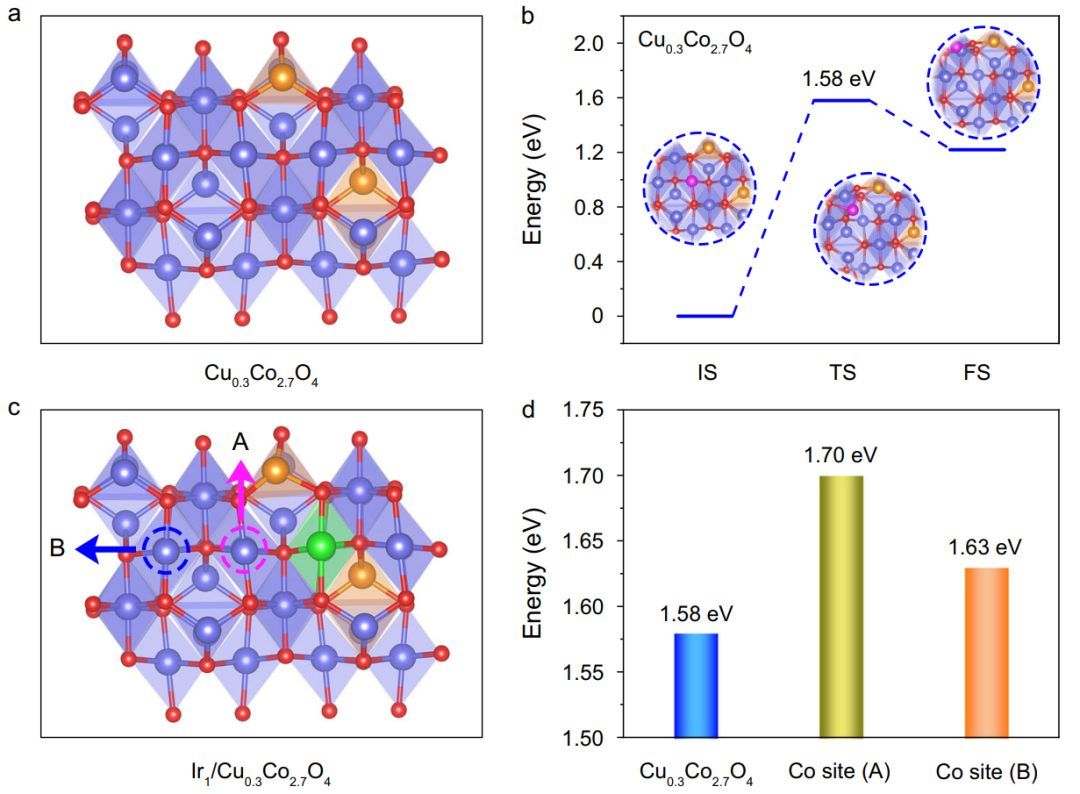
从DFT计算开始,研究了引入耐酸杂原子如何影响尖晶石氧化物中Co原子的稳定性。由于钴氧化物在酸中的溶解涉及到钴原子在表面的迁移,计算了这些晶格原子的迁移能来评估它们的稳定性。以尖晶石Cu0.3Co2.7O4为模型(图1a),提出了Co原子在八面体位点的迁移过程,如图1b所示,其中Co原子通过一个能量势垒为1.58 eV的过渡态离开(110)面。在将Ir单原子引入氧化尖晶石的八面体位点后(图1c),首先计算了最近的Co原子(标记为A)到Ir单原子的迁移能。 随后,进一步评估了Ir单原子对远处Co原子迁移的影响,即图1c中标记为B的下一个最近的Co原子。计算得到B位点Co的迁移能为1.63 eV ,说明在不引入Ir的情况下,B位点Co原子比A位点Co原子稳定,但比Co原子稳定(图1d)。
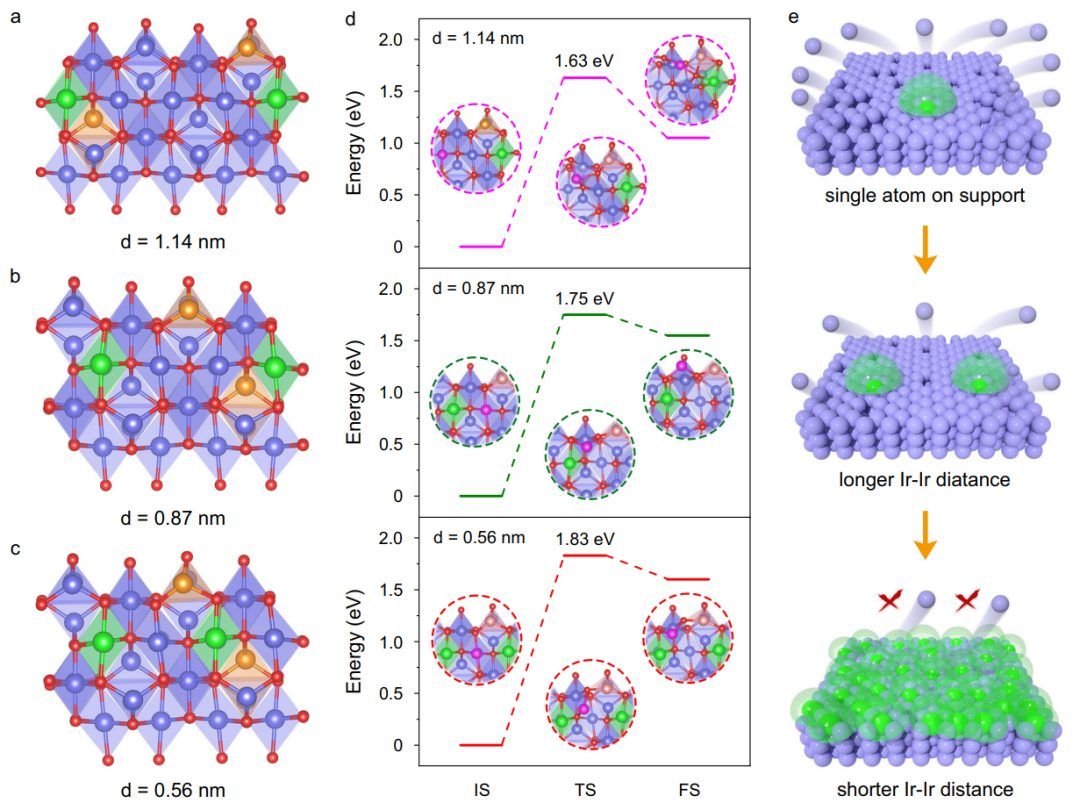
为了进一步探索Ir单原子的距离依赖稳定效应,构建了不同Ir-Ir距离的Ir原子掺杂尖晶石钴氧化物Ir1/Cu0.3Co2.7O4的各种结构模型(d)。图2a-c显示了d=1.14、0.87和0.56 nm的情况,其中相邻两个Ir原子之间的Co原子数分别为3、2和1。当d = 1.14 nm时,计算出居心Co原子的迁移能为1.63 eV。当d = 0.87 nm时,能量为1.75 eV(图2d)。 迁移能的增加表明,相邻Ir单原子之间的距离越短,催化剂表面的Co原子越稳定。上述结果表明,Ir单原子可以稳定邻近晶格,但对远处晶格的影响有限。当相邻Ir单原子之间的距离过大时,Ir单原子的稳定作用非常有限。一旦Ir单原子之间的距离减小到一定值,稳定作用就会有效地覆盖尖晶石氧化物,从而在酸性条件下稳定整个尖晶石氧化物。
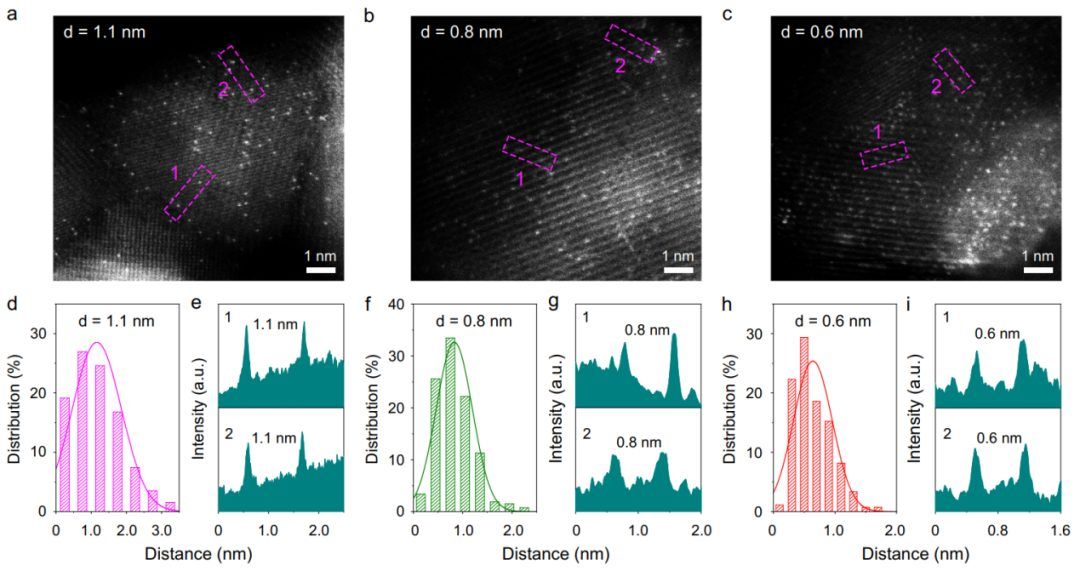
图3a-c为三个不同Ir-Ir距离的Ir1/Cu0.3Co2.7O4样品经像差校正后的HAADF-STEM图像。与Cu0.3Co2.7O4相比,发现了个别亮点,这表明Ir原子分散在尖晶石钴氧化物中。此外,通过平均HAADF-STEM图像中200多个Ir-Ir原子对的Ir-Ir距离,估计d的值分别约为1.1,0.8和0.6 nm(图3d-i)。

本研究利用XANES和EXAFS进一步表征了不同Ir-Ir距离的Ir1/Cu0.3Co2.7O4的电子结构和配位环境。Co的K边XANES光谱显示,不同Ir-Ir距离的Ir1/Cu0.3Co2.7O4的吸收边与Cu0.3Co2.7O4的吸收边重叠,表明所有氧化物中Co的价态相似。Co的K边EXAFS光谱在约1.4、2.3和3.0 Å处呈现3个相似的特征峰,分别对应于Co-O、Cooct-Cooct(八面体位点)和Cotet-Cotet(四面体位点)(图4b),说明引入单原子Ir后Co位点的配位环境没有明显变化。 上述结果表明,引入不同距离的Ir单原子后,钴氧化物的晶体结构和电子结构没有明显的变化。图4c为Ir1/Cu0.3Co2.7O4在不同Ir-Ir距离下的Ir的L3边缘XANES光谱。白线的强度随着Ir-Ir距离的减小而逐渐减小,表明Ir的价态降低。Ir的L3边EXAFS光谱在2.0 Å和3.0 Å处表现出两个特征峰,分别属于第一壳层Ir-O配位和第二壳层Ir-Co配位(图4d)。通过对实验EXAFS光谱的拟合,确定了不同Ir-Ir距离下Ir1/Cu0.3Co2.7O4的Ir-O配位数和Ir-Co配位数分别约为6和3。拟合结果证实了Ir单原子被引入Cu0.3Co2.7O4的八面体位点。
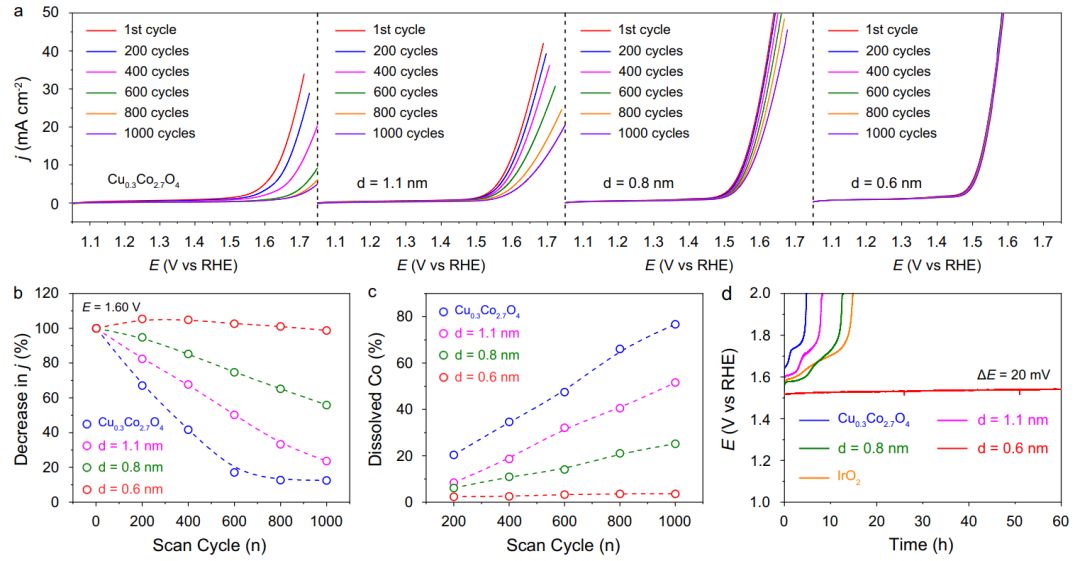
为了评价不同距离的Ir单原子对Cu0.3Co2.7O4的稳定作用,记录了标准三电极体系在酸性介质下的极化曲线。对于原始Cu0.3Co2.7O4,随着扫描次数的增加,电流密度明显降低(图5a)。经过1000次扫描循环后,电流密度下降了87.5%。在Cu0.3Co2.7O4中引入d=1.1和0.8 nm的Ir单原子后,经过1000次扫描循环,电流密度分别下降了76.3%和44.2%(图5a、b)。当Ir-Ir距离进一步减小到0.6 nm时,在连续扫描过程中电流密度下降不明显,表明该样品在酸性介质中具有良好的OER稳定性(图5a、b)。 本文还测量了不同扫描次数下Co物质的溶解情况,探讨了不同Ir-Ir距离下Cu0.3Co2.7O4和Ir1/Cu0.3Co2.7O4的稳定性。Cu0.3Co2.7O4中的Co随扫描次数的增加而逐渐溶解。具体来说,在1000次扫描循环后,76.7%的Co物种被溶解(图5c)。当在Cu0.3Co2.7O4中引入Ir=1.1和0.8 nm时,Co的溶解速度减慢,说明Ir单原子可以稳定Cu0.3Co2.7O4。当在Cu0.3Co2.7O4中引入Ir=0.6 nm时,经过1000次扫描循环后,Co仅溶解(仅3.6%),表明d=0.6 nm时Cu0.3Co2.7O4具有较高的稳定性。 在恒电流密度为10 mA cm-2geo的条件下,用长期计时电位法测定了不同Ir-Ir距离下Cu0.3Co2.7O4和Ir1/Cu0.3Co2.7O4的耐久性。如图5d所示,这些催化剂的耐久性随着Ir-Ir距离的减小而提高。当d=0.6 nm时,连续工作60 h后,催化剂保持稳定,电势仅小幅增加约20 mV。值得注意的是,与商业IrO2相比,d=0.6 nm的Ir1/Cu0.3Co2.7O4具有更好的稳定性。上述结果表明,Ir单原子诱导的稳定效应强烈依赖于相邻单原子的距离。
文献信息
Distance effect of single atoms on stability of cobalt oxide catalysts for acidic oxygen evolution,Nature Communications,2024.
2 JACS:拓扑催化CO2电还原的实验验证

近年来,拓扑物理学被进一步扩展到化学学科,导致拓扑催化的出现。原则上,拓扑效应在催化反应中是可检测到的,但尚未有确凿的证据报道。 中国科学技术大学曾杰教授、王征飞教授等人通过厚度控制和磁场的应用,精确控制Bi2Se3纳米片的拓扑表面态(TSS),为说明拓扑催化CO2电还原提供了直接的实验证据。在TSS的配合,CO2主要还原为液体燃料(HCOOH和H2C2O4),表现出较高的法拉第效率(FE)(在-1.1 V下可达90%);而缺乏TSS时,CO2主要还原为CO。理论研究表明,产物和FE的差异可归因于TSS调节的关键中间体的吸附和电位决定步骤的降低。该工作证明了带拓扑结构与电催化之间的内在相关性,为设计高性能催化剂铺平了新的道路。
相关工作以《Experimental Demonstration of Topological Catalysis for CO2 Electroreduction》为题在《Journal of the American Chemical Society》上发表论文。
图文导读
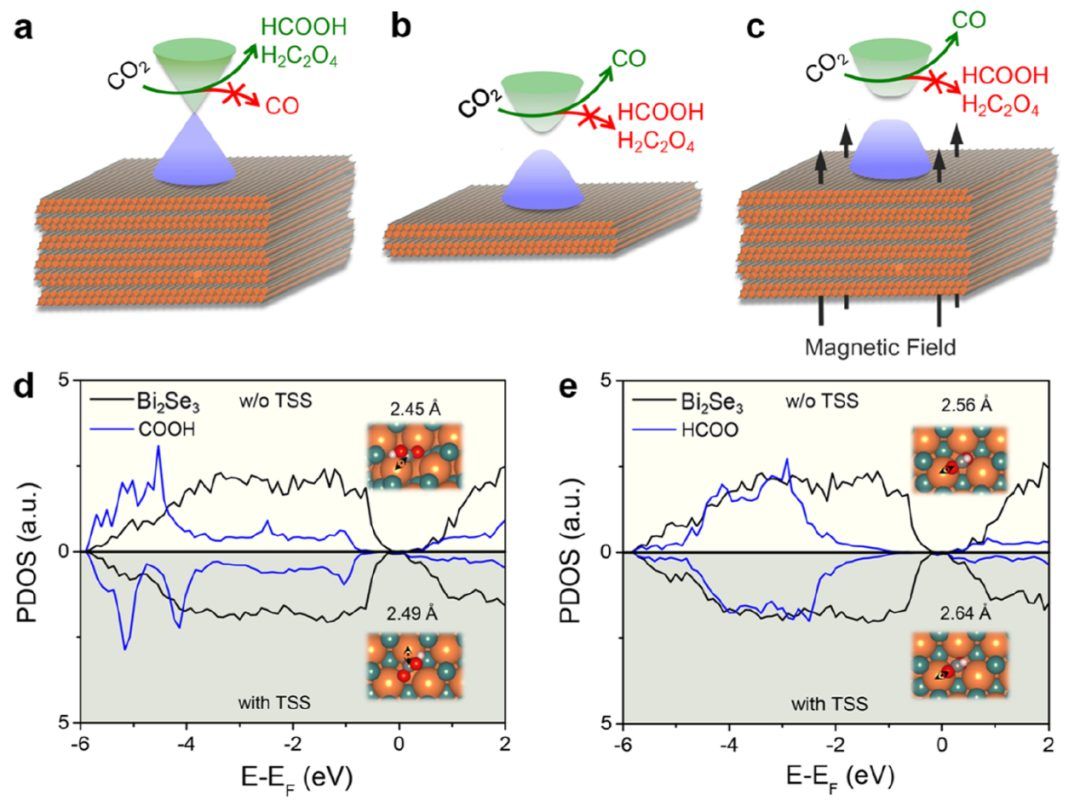
本文设计了三个互补的实验方案来揭示神秘的拓扑催化CO2电还原。在第一个实验中(图1a),选择了带有TSS的厚Bi2Se3。在第二个实验中(图1b),选择了没有TSS的薄Bi2Se3,其中TSS被量子限制消除了。在第三个实验中(图1c),选择了较厚的Bi2Se3,但在磁场下进行催化反应,其中时间反转对称保护的TSS被磁场破坏。显然,如果第二和第三实验中的催化活性或主导产物在第一实验中相似但不同,则可以直接确认内在的TSS效应是调节催化活性或选择性。 在进行电化学测量之前,在4个五层(4-QL)Bi2Se3上进行了*COOH和HCOO*第一性原理计算,以确定TSS对CO2电还原中间体的吸附是否有显著影响。*COOH和HCOO*的PDOS分别如图1d、e所示。比较有TSS和没有TSS的PDOS,可以看到TSS使*COOH和HCOO*的占据态向费米能级上升,表明吸附行为减弱。计算得到的键长和吸附能(Ead)也支持了这一结论。对于*COOH的吸附,TSS使Bi-C键增加0.04 Å,使Ead减少0.07 eV。因此,Bi2Se3的TSS对中间体的吸附有相当大的影响,从而可以通过实验检测TSS调节的催化活性和选择性。
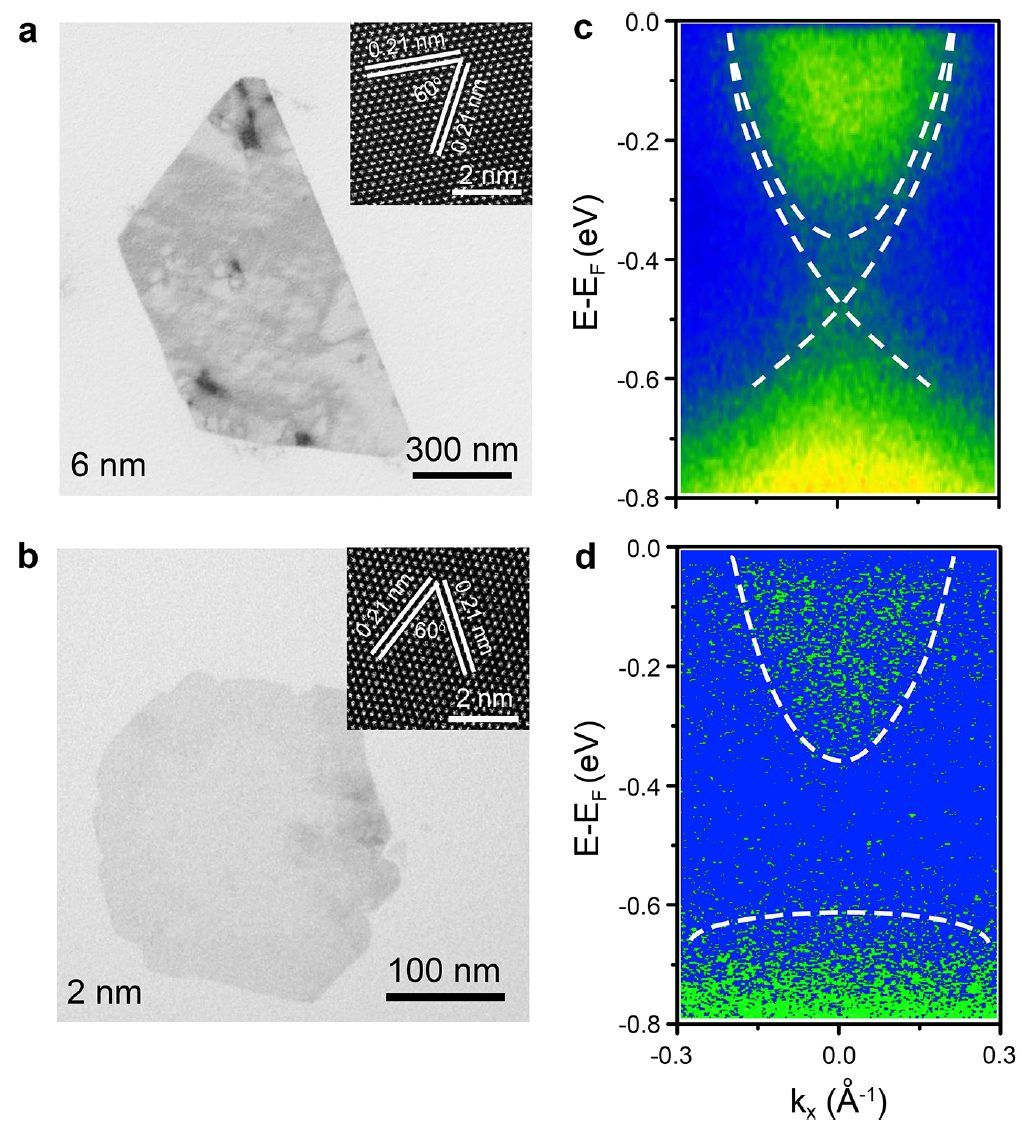
6 nm和2 nm的Bi2Se3纳米片通过TEM(图2a、b)和HAADF-STEM(插图)图像来表征,显示高质量的单晶具有六方晶结构。为了直接鉴定制备样品中的TSS,进行了角分辨光电子能谱(ARPES)测量。在ARPES光谱中,6 nm Bi2Se3纳米片在费米能级以下表现出狄拉克锥的TSS(图2c),其能量分布曲线(EDC)的光谱峰位于Γ点,而2 nm Bi2Se3纳米片在费米能级以下表现为间隙态(图2d)。
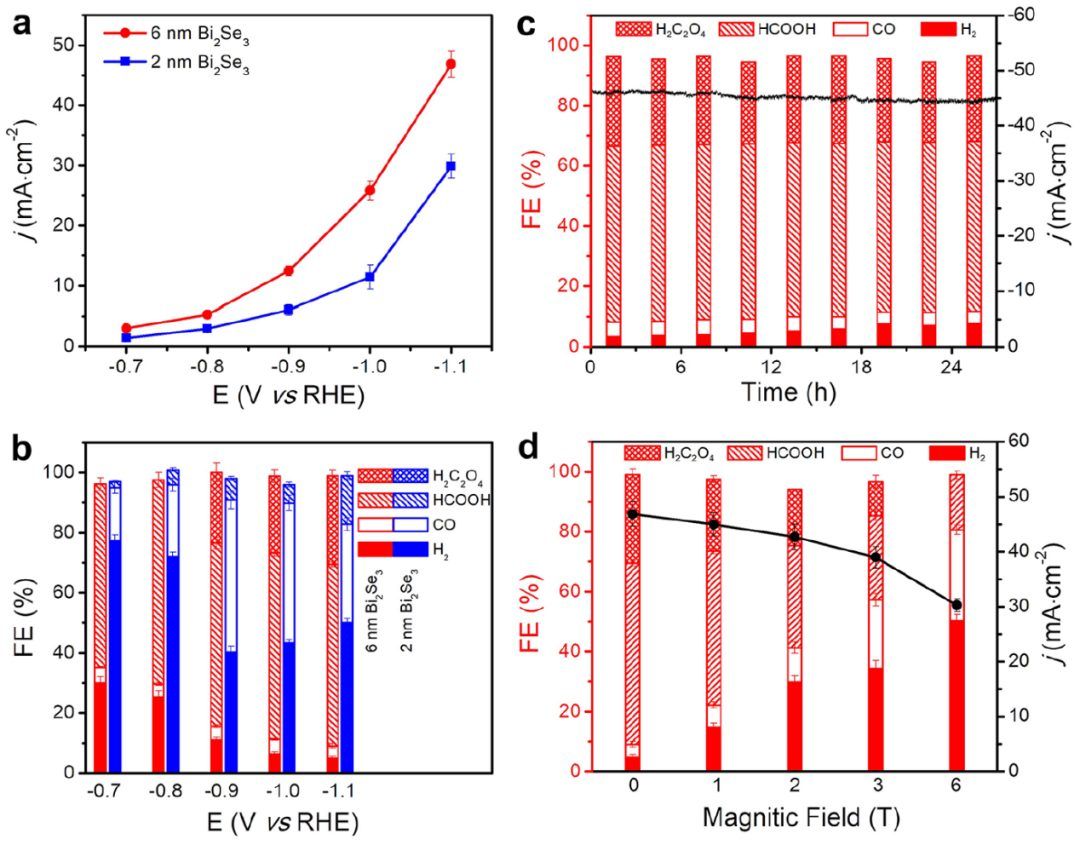
图3a显示了6 nm和2 nm Bi2Se3纳米片上CO2电还原的几何电流密度(j)。j随电位的增加而单调增加,其中6 nm Bi2Se3纳米片在所有电位下都显示出较大的值。在-1.1 V下,6nm Bi2Se3纳米片的j甚至增加到46.9 mA·cm-2,是2 nm Bi2Se3纳米片(29.9 mA·cm-2)的1.6倍。 除j外,CO2电还原产物的分布对Bi2Se3纳米片的厚度也很敏感。在测量中可以观察到三个显著的特征,如图3b所示。(1)6 nm的Bi2Se3纳米片的主要产物是液体燃料(HCOOH和H2C2O4),在所有电位下都具有较高的FE,而2 nm的Bi2Se3纳米片的主要产物是CO,在所有电位下都具有较低的FE。
对于6 nm的Bi2Se3纳米片,HCOOH存在于所有电位,而H2C2O4只存在于高电位(-0.9、-1.0和-1.1 V)。(3)对于2 nm的Bi2Se3纳米片,HCOOH在所有电位下都存在,但H2C2O4不存在。 为了测试6 nm的Bi2Se3纳米片的稳定性,在-1.1 V下进行了稳定性测量。在连续反应27 h时,j的衰减小于6%,HCOOH和H2C2O4的FE值分别保持在56%和27%以上(图3c)。此外,6 nm的Bi2Se3纳米片上在磁场下原位进行CO2电还原。在-1.1 V下,随着磁场强度从0增加到6 T(图3d),j从46.9 mA·cm-2减小到30.3 mA·cm-2。同时,液体燃料的FE从90% (HCOOH和H2C2O4)降低到18% (HCOOH)。显然,通过施加磁场,活性降低和主导产物(从液体燃料到CO)的改变使6 nm的Bi2Se3纳米片更类似于2 nm的Bi2Se3纳米片。

图4a-c分别为添加TSS和不添加TSS的4-QL的Bi2Se3上CO、HCOOH和H2C2O4生成的吉布斯自由能图。添加TSS的Bi2Se3生成*COOH的ΔG为0.40 eV,比未添加TSS的Bi2Se3(0.26 eV)高0.14 eV。相比之下,有TSS(0.13 eV)的HCOO*生成的ΔG比没有TSS (0.53 eV)的低0.40 eV。对于H2C2O4的生成,考虑了CO2与*COOH偶联(路径III)和共吸附*COOH之间的偶联两种反应路径。如图4c所示,经计算,在有TSS的Bi2Se3上生成H2C2O4*的ΔG为0.56 eV,比没有TSS的Bi2Se3(0.64 eV)低0.08 eV。因此,含有TSS的Bi2Se3进行CO2电还原时,液态产物HCOOH和H2C2O4比CO更有利。形成H2C2O4的PDS最高ΔG表明,在6 nm的Bi2Se3纳米片上形成H2C2O4需要更高电位。 本研究进一步采用目标产物和副产物的热力学极限电位差(定义为ΔUL=UL(目标产物)-UL(副产物),其中UL=-ΔG/e(ΔG为PDS的能垒))来评价产物选择性。ΔUL值越正,对应于目标产物的选择性越高。图4d显示了HCOOH和CO之间的ΔUL关系,在添加和不添加TSS的Bi2Se3中,ΔUL分别为0.27 V和-0.23 V。图4e显示了H2C2O4和CO之间的ΔUL,分别为-0.16 V和-0.34 V。在这两种情况下,有TSS的ΔUL比没有TSS的ΔUL更正,进一步支持TSS调节CO2电还原活性和选择性的事实。
文献信息
Experimental Demonstration of Topological Catalysis for CO2 Electroreduction,Journal of the American Chemical Society,2024.10.1021/jacs.3c11088