基因检测为什么瞄准耐药性
根据《肿瘤的发生与进展背后的基因机制》,卵巢癌是妇科癌症死亡的首要原因,耐药性是卵巢癌治疗的瓶颈。随着新型药物在临床上的不断应用,耐药性卵巢癌的治疗成为新的关注重点,这不是因为治疗更困难,而是卵巢癌的治疗向前推进了一大步。继续按照药物类型对耐药性进行分类而不了解其潜在机制并不满足更高治疗水准的要求。肿瘤耐药性基因检测回顾了卵巢癌各种耐药机制的基因解码证据,揭示了卵巢癌的耐药机制:跨膜转运异常、DNA损伤修复改变、癌症相关信号通路失调和表观遗传修饰。DNA甲基化、组蛋白修饰和非编码RNA活性是三大关键的表观遗传修饰,是耐药的关键机制。一种药物可以有多种耐药机制。此外,常见的化疗和靶向药物可能存在交叉(重叠)的耐药机制。微小RNA(miRNA)可以干扰并调节上述通路。 miRNA 的一个类型,在基因解码中被称为“epi-miRNA”的基因信息载体,可以调节表观遗传调节因子,从而影响治疗反应。因此,卵巢癌的耐药性的基因解码团队总结了 miRNA 对抗性机制的调节影响。此外,卵巢癌的耐药性基因检测团队还总结了基于上述抗性机制的卵巢癌新药的近期 I/II 期临床试验。许多新疗法正在接受评估,初步结果令人鼓舞。《卵巢癌的基因解码基因检测》为卵巢癌药物抗性机制的分类提供了新的见解,并可能有助于成功治疗抗性卵巢癌。
卵巢癌基因检测关键词:
卵巢癌,miRNA,耐药机制,临床试验
妇科肿瘤基因检测项目对卵巢癌的治疗与耐药性的地平线介绍
卵巢癌 (OC) 是女性生殖系统第三大常见且最致命的恶性肿瘤。70% 的患者在确诊时已处于晚期 (FIGO III 期和 IV 期) 且存在远处转移。尽管接受了标准治疗(最佳细胞减灭手术后进行辅助化疗),但大多数患者会出现复发,并且对化疗具有耐药性,导致全球 5 年生存率约为 30–40%。虽然使用聚(腺苷二磷酸核糖)聚合酶 (PARP) 抑制剂 (PARPis) 进行维持治疗可延长无进展生存期 (PFS) 和 5 年总生存期 (OS) ,但不幸的是,许多患者由于内在或获得性耐药而对 PARPi 治疗没有反应。耐药性是卵巢癌治疗的一大挑战,也是导致预后不良的主要原因。
根据美国国家综合癌症网络(NCCN)指南(1.2023版),治疗耐药性卵巢癌的方案很多,包括一些新型药物。然而,由于耐药机制复杂,客观缓解率仍然较低,中位生存期不足12个月。在耐药性卵巢癌中,常用药物的经典作用机制可能被打乱或改变,从而可能导致治疗效果受损。因此,治疗方案不应仅依赖经验选择。除传统药物外,新型化合物也正在早期临床试验中进行研究和测试。随着药物种类的增加,在出现多药耐药(MDR)后,选择合适的后线治疗方案非常具有挑战性。这个问题促使佳学基因检测考虑不同药物之间耐药机制的相互作用。
即使对不同的药物会产生耐药性,其潜在机制可能相似。因此,在找出耐药性原因的过程中,基因解码师没有简单地根据药物区分耐药性,而是尝试根据机制对耐药性进行分类。肿瘤的耐药性基因检测基因解码团队从已发表的文献中总结了四种主要机制(图 1):1)跨膜转运异常,2)DNA 损伤修复(DDR)改变,3)癌症相关信号通路失调,4)表观遗传修饰。微小 RNA(miRNA)在转录后调节靶基因的表达并影响多种生物过程,包括癌细胞增殖、转移和治疗耐药性。miRNA 通过作用于与上述四种机制相关的分子或/和通路来显著调节耐药性。miRNA 表达异常可导致控制药物流入和流出的药物转运蛋白失调。 DDR 机制中某些成分的表达,如同源重组修复 (HRR) 和非同源末端连接 (NHEJ),受到 miRNA 的调节。此外,miRNA 可以通过靶向多种癌症相关信号通路的成分来干扰这些通路,从而促进肿瘤对治疗产生耐药性。
图 1.miRNA介导的耐药机制概述
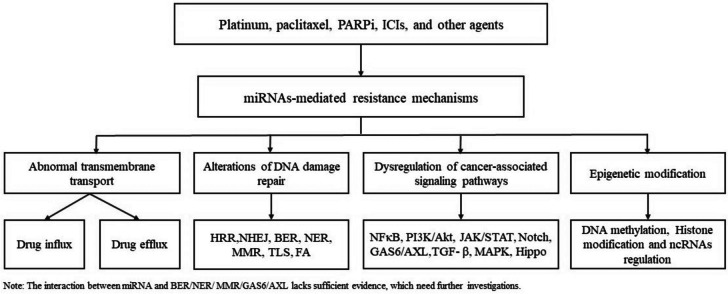
图1: miRNA介导的耐药机制概述 ( a ) 跨膜转运异常;( b ) DNA损伤修复改变;( c ) 癌症相关信号通路失调;( d ) 表观遗传修饰
基于上述发现,卵巢癌耐药性基因检测检索了用于治疗卵巢癌耐药的新药的 I/II 期临床试验(表 1 )。了解潜在的耐药机制有望有助于发现逆转耐药的新临床选择并改善卵巢癌患者的预后。
表 1.基于机制的卵巢癌耐药性 I/II 期临床试验总结。
I/II 期临床试验中的一些新药试图通过靶向跨膜转运、DNA 损伤修复、信号通路和表观遗传修饰来逆转卵巢癌的耐药性。卵巢癌耐药性基因检测总结了临床试验的这些组成部分,包括研究标识符、阶段、试验药物、涉及的靶点、疾病状况、主要结果指标、研究状态、研究结果
临床实验编号
阶段
实验药物
针对靶点
疾病状况
主要考察指标
试验状态
研究结果
针对跨膜转运的临床试验
NCT04918186
II
BA3011(BA3021)+Durvalumab
Axl (ROR2)
铂类耐药性高级别浆液性卵巢癌
客观缓解率
招募
未获得
NCT01335958
I
DMUC5754A
MUC16
铂类耐药性卵巢癌
DLT 系统
已完成、已发布
≥5% 的≥3 级相关不良事件,在 MUC16 高患者中具有抗肿瘤活性
NCT02146313
I
DMUC4064A
MUC16
铂类耐药性卵巢癌
DLT、MTD、PR2D、AE、sAE
已完成 发布
25% 的患者发生 3 级以上毒性反应
RP2D:5.2 毫克/千克,CBR:MUC16 高患者中为 46%
NCT01469793
I
DMOT4039A
Mesothelin
铂类耐药性卵巢癌
MTD、DLT RP2D
已完成,已出版
RP2D: 2.4毫克/千克(每三周一次)和1.0毫克/千克(每一周一次); SAE:8.5%
NCT02751918
Ib
BAY94-9343+PLD
Mesothelin
表达间皮素的铂类耐药性复发性卵巢癌、输卵管癌或原发性腹膜癌
MTD、AE
已完成 发布
ORR:27.7%(全部) ORR:42.1%(高间皮素表达) MTD:6.5 mg/kg
最常见的不良反应:恶心(47.7%)
NCT01363947
I
DNIB0600A
NaPi2b
非粘液性及铂类耐药性卵巢癌
AE、DLT、RP2D、OR、DOR
已完成 发布
≥3级中性粒细胞减少症(10%)RP2D:2.4 mg/kg(q3w)
所有 RECIST 反应 (NaPi2b-high)
NCT04504916
II
齐洛他单抗
ROR1
铂类耐药性卵巢癌
ORR、TTR、DOR、PFS、OS
完全的
未获得
NCT02539719
1a/1b
Tamrintamab 帕莫西林
DPEP3
铂类耐药/难治性卵巢癌
不良事件、ORR
已完成 发布
ORR:4%(无法忍受)DPEP3 越高,反应越好
针对 DDR 的临床试验
NCT02595892
II
盐酸吉西他滨 + M6620
ATR
复发性、铂类耐药性高级别浆液性卵巢癌
PFS、ORR
已完成 发布
展示将 M6620 添加到吉西他滨的一些益处(PFS:22.9w vs 14.7w)
NCT04149145
I
M4344+尼拉帕尼
ATR
PARPi 耐药晚期上皮性浆液性卵巢癌、原发性腹膜癌或输卵管癌
AE、MTD
尚未招募
未获得
NCT03462342
I
AZD6738+ 奥拉帕尼
ATR
复发性铂敏感和铂耐药性 HGSOC
不良事件、ORR、PFS
招募
未获得
NCT03704467
Ib//II
卡铂 + M6620 + 阿维单抗
ATR
PARPi 抗性卵巢癌
DLT、AE
招募已完成
未获得
NCT01164995
II
MK-1775 + 卡铂
WEE1
患有 TP53 突变的难治性或铂类耐药性卵巢癌
不良事件、抗肿瘤活性 (CT/CA125)
已完成 发布
ORR:41%,PFS:5.6m AE:骨髓毒性、恶心和呕吐
NCT03579316
II
AZD1775+奥拉帕尼
WEE1
PARPi 抗性卵巢癌
ORR、安全性和耐受性
招募
未获得
NCT02272790
II
Adavosertib+卡铂/
WEE1
铂类耐药性上皮性卵巢癌、输卵管癌或原发性腹膜癌
ORR、不良事件
已完成 发布
ORR(总体):31.9% ORR(adavosertib+卡铂):66.7% ≥3 级不良事件:贫血(33%)、中性粒细胞减少症(45.7%)、血小板减少症(31.9%)
PLD/紫杉醇/
吉西他滨
NCT05198804
I/II
ZN-c3 + 尼拉帕尼
WEE1
铂类/PARPi 耐药性卵巢癌
DLT、PFS、ORR
招募
未获得
NCT04516447
I
ZN-c3+ PLD/卡铂/紫杉醇/吉西他滨
WEE1
铂类耐药性卵巢癌、腹膜癌或输卵管癌
AE、MTD、RP2D
招募
未获得
NCT02101775
II
吉西他滨联合或不联合 MK-1775
WEE1
复发性、铂类耐药性上皮性卵巢癌、原发性腹膜癌或输卵管癌
PFS、OR、OS、AE
已完成 发布
PFS:4.6 个月 vs 3.0 个月(HR 0.56,95%CI:0.35 至 0.90,p =0.015)。OS:11.5 个月 vs 7.2 个月(HR 0.56,95%CI 0.34 至 0.92,p =0.022)。PR 率:21% vs 3%(p =0.02)
NCT02203513
II
LY2606368
CHK1/2
复发性铂类耐药 HGSOC,伴有 BRCA 野生型或突变
客观缓解率
部分完成(BRCA 宽型),招募正在进行中
PR(按方案可评估):33%(8/24),≥3 级 AE:中性粒细胞减少症(93%);白细胞计数减少(82%);血小板减少症(25%)、贫血(11%)。
NCT03414047
II
LY2606368
CHK1/2
具有 BRCA 野生型或突变的铂类耐药 HGSOC
客观缓解率
完全的
在铂类耐药患者中:ORR(队列 1-3):12.1% DCR 为 37.1%,
NCT04678102
I
PHI-101
CHK2
铂类耐药性卵巢癌、原发性腹膜癌或输卵管癌
DLT、MTD
招募
未获得
NCT02797964
I/II
SRA737
CHK1
耐铂或不耐铂 HGSOC
AE、MTD
完全的
最大耐受剂量:1000 毫克,每日一次
RP2D:800 毫克每日一次
轻微毒性
针对信号通路的临床试验
NCT03875820
I
德法替尼+VS-6766
MAPK
未经常规治疗的 LGSOC
确定耐受剂量并测量。不良事件
活跃,不招募
未获得
NCT03648489
II
TAK228
PI3K/AKT/mTOR
铂类耐药性卵巢癌
增强体质
活跃,不招募
未获得
NCT03586661
I
科潘利斯布
PI3K/AKT
伴有 BRCA 突变的铂类耐药性卵巢癌
MTD 和 RP2D
活跃,不招募
未获得
NCT04374630
II
阿呋塞替+紫杉醇
PI3K/AKT
铂类耐药性卵巢癌
增强体质
招募
未获得
NCT04586335
I
CYH33
PI3K/AKT
铂类/PARPi 耐药性卵巢癌
DLT,ORR
招募
未获得
NCT04055649
II
ONC201
PI3K/AKT、MAPK
铂类难治性或耐药性卵巢癌
不良事件、DLT、ORR、PFS
招募
未获得
NCT05295589
II
科潘利斯布
PI3K/AKT
复发性铂类耐药性卵巢癌
增强体质
尚未招募
未获得
NCT03363867
II
考比替尼
MAPK
复发性铂类耐药性高级别浆液性卵巢癌
客观缓解率
招募
未获得
NCT03639246
I/II
AVB-S6-500 型
GAS6-AXL
铂类耐药性复发性卵巢癌
不良事件
已完成 发布
ORR (AVB-500 + PAC):34.8% 中位 DoR、PFS 和 OS (AVB-500 + PAC):分别为 7.0、3.1 和 10.3 个月 RP2D (AVB-500):15 毫克/千克
增强体质
NCT04019288
I/II
AVB-S6-500 型
GAS6-AXL
铂类耐药性卵巢癌
不良事件
已完成 发布
6 周内无 DLT 和 ≥3 级 AE 正在进行探索性研究。
NCT04893551
I
替维司他单抗
GAS6-AXL
铂类耐药复发性 HGSOC
不良事件
已终止
未获得
NCT01952249
Ib
地西珠单抗+紫杉醇
Notch
铂类耐药性卵巢癌、原发性腹膜癌和输卵管癌
DLT 系统
完全的
RP2D:3.5毫克/千克
耐受性、临床活动、
NCT03776812
II
瑞科瑞兰 +
GR
复发性铂类耐药性卵巢癌、输卵管癌或原发性腹膜癌
PFS、ORR、DOR
完全的
各组间的 ORR 相似 (35%);间歇组:
白蛋白紫杉醇
OS,13.9个月,PFS,5.6个月连续组;
OS,11.3 个月,PFS,5.3 个月 Nab-紫杉醇:OS,12.2 个月,PFS,3.8 个月
NCT03319628
1/II
XMT-1536
NaPi2b
铂类耐药性卵巢癌
MTD 和 RP2D
招募
未获得
NCT04502602
1/1b
尼拉帕尼 + 来那替尼
HER2
铂类耐药性卵巢癌
RP2D、PFS、AE
招募
未获得
NCT03287271
I/II
Defactinib (VS-6063) +卡铂/紫杉醇
FAK
卡铂耐药性卵巢癌
ORR、不良事件
招募
未获得
针对表观遗传修饰的临床试验
NCT05327010
II
ZEN003694 +他拉唑帕尼
BET bromodomain
PARPi 耐药复发性卵巢癌,伴有 BRCA 突变或 DDR 畸变
客观缓解率
招募
未获得
NCT04840589
I
ZEN003694+nivolumab+/-伊匹木单抗
BET bromodomain
复发性铂类耐药性卵巢癌
RP2D
招募
未获得
NCT03206047
I/II
阿替利珠单抗+/-Guadecitabine+/-CDX-1401 疫苗
DNMT
铂类耐药性上皮性卵巢癌、输卵管癌或原发性腹膜癌
不良事件、无进展生存期
招募
未获得
NCT02901899
II
Guadecitabine + 帕博利珠单抗
DNMT
复发性铂类耐药卵巢癌
客观缓解率
完全的
PR:8.6%;标准差:22.9%; CBR:31.4%(95% CI:16.9%-49.3%);
临床获益持续时间为 6.8 个月
ORR客观缓解率,DLT剂量限制性毒性,MTD最大耐受剂量,PR2D推荐 II 期剂量,AE不良事件,sAE严重不良事件,PFS无进展生存期,CBR临床受益率,GR糖皮质激素受体,DoR缓解持续时间
卵巢癌耐药机制
跨膜转运异常
跨膜转运异常的两种形式是药物流入减少和流出增加,这会导致细胞内药物浓度降低并产生耐药性(图 2 )。此外,在铂类耐药的卵巢癌( PROC )中,相关基因和转运蛋白的表达降低。因此,细胞内铂类药物浓度不足,随后产生铂类耐药性。miRNA 可以直接靶向跨膜转运蛋白,从而调节细胞对药物的耐药性。它们直接与靶转运蛋白基因的 3'-非翻译区(3'-UTR)结合以调节其转录,导致药物流入和流出异常 。
图 2.跨膜转运异常
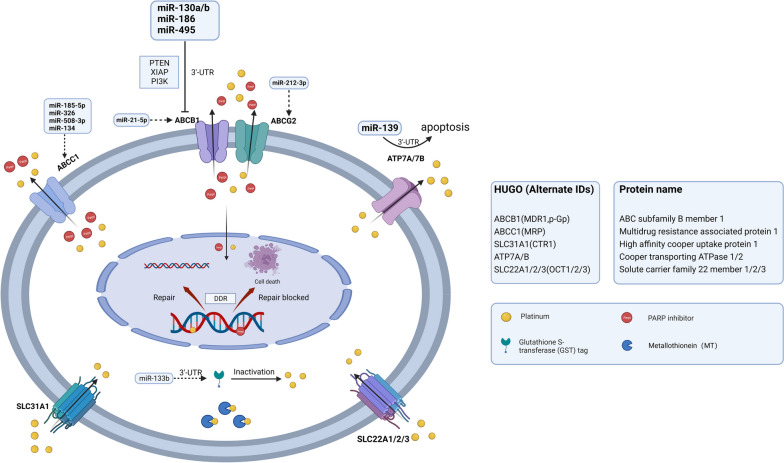
图2:跨膜转运异常。SLC31A1、SLC22A1/2/3作为SLC超家族成员,是负责药物内流的重要转运体。SLC转运体下调使铂类药物的摄取减少,导致卵巢癌产生化疗耐药。佳学基因检测仍然在丰富miRNA在SLC表达中的作用的证据。ABC转运体家族包括ABCB1、ABCG2、ABCC1,负责药物外排,从而降低细胞内铂类药物浓度。miR130a/b、miR-186、miR-495可直接与ABCB1 mRNA的3’-UTR结合或调控PTEN、XIPA、PI3K,导致ABCB1转录或翻译水平下降。miR-21-5p和miR-212-3p也分别对ABCB1和ABCG2有调控作用。 miR-185-5p、miR-326、miR-508-3p和miR-134可以调控ABCC1的表达。ATP7A/7B是药物外排的另一个贡献者。miR-139可以直接与ATP7A/7B的3'-UTR结合,导致细胞凋亡诱导并增加卵巢癌细胞的化疗敏感性。MT可以与顺铂结合并使其失活,从而降低药物疗效并诱导耐药性。GST催化谷胱甘肽与铂结合并导致药物失活,这与卵巢癌的铂耐药性有关。(SLC,溶质载体超家族;GST,谷胱甘肽转移酶;MT,金属硫蛋白)
减少药物流入
细胞膜或质膜上的钠泵、铜离子转运蛋白和有机阳离子转运蛋白,如药物转运溶质载体 (SLC) 超家族转运蛋白(如 SLC31A1、SLC22A1/2/3),是控制药物内流的关键转运蛋白。已确凿证明 SLC31A1 能转运顺铂及其类似物卡铂和奥沙利铂,导致铂在细胞内蓄积。卵巢癌中 SLC22A2 的低表达可能与铂类药物耐药性相关,这是因为铂类药物的摄取减少。miRNA 在药物转运 SLC 转运蛋白的表达中起着关键作用,可能影响前列腺癌、肝细胞癌和结直肠癌的治疗反应。然而,miRNA 与 SLC 转运蛋白在卵巢癌耐药性中的关联和相互作用机制仍需要基因解码进一步丰富,以便提供更有价值的基因解码基因检测结果。
药物外排增加
ABC转运蛋白家族主要负责药物外排。miRNA(如miR-200家族、let-7家族和miR-130a/b)的异常表达在ABC转运蛋白的调控中发挥作用,从而诱导卵巢癌的耐药性。ABC家族中已鉴定的外排转运蛋白包括ABCB1、ABCG2和ABCC。上述miRNA可以与编码ABC转运蛋白的mRNA的3’-UTR结合,或参与与编码核受体、转录因子(TF)和与ABC转运蛋白相关的信号分子的基因进行不完全碱基配对。通过这种作用,ABC转运蛋白的mRNA被降解或相应蛋白质的翻译受到抑制。此外,穹窿蛋白肺耐药相关蛋白(LRP)可以将细胞抑制药物从细胞内靶点转运,从而产生耐药性。
全基因组微阵列分析显示,ABCB1是唯一一种在耐药卵巢癌细胞中表达增加的药物转运蛋白,而其他几种 ABC 转运蛋白的表达则显著下降。膜转运蛋白 P-糖蛋白 (P-gp) 由ABCB1编码,是一种 ATP 依赖性药物外排泵。其在耐药细胞系中的过表达被认为是对紫杉醇、阿霉素、索拉非尼和 PARPis产生耐药性的关键机制。值得注意的是,失调的 miRNA 可以介导 ABCB1 的过表达,从而导致 MDR。例如,miR130a/b、miR-186 和 miR-495 可以直接结合 ABCB1 mRNA 的 3'-UTR 或调节其他靶标(如 PTEN、XIAP 和 PI3K)的表达,导致 ABCB1 mRNA 降解或翻译抑制。研究发现,ABCB1 表达的大幅增加与 miR-21-5p 表达的降低相关,但所涉及的调节机制仍然未知。此外,ABCB1的上调与 ABCB1 和 SLC25A40 的转录融合有关,这种融合是通过在接受过化疗和靶向治疗的高级别浆液性卵巢癌 (HGSOC) 患者中进行的全基因组分析发现的。这些研究结果表明,ABCB1 上调常常会诱导对化疗药物和靶向药物的交叉耐药性。因此,不依赖 P-gp 转运蛋白的 PARPis 可能对接受过化疗的患者表现出更高的治疗效果。ABCC1 与 HGSOC 中的较差生存率和化学耐药性有关。miR-185-5p 和 miR-326 都靶向 ABCC1 3'-UTR 来调节 ABCC1 的表达。miR-508-3p和 miR-134分别被 CircETDB1 和 LINC01118 吸收,可以在转录后调节 ABCC1 的表达。ABCG2 参与卵巢癌的拓扑替康耐药性,这与 miR-212-3p 下调有关。
此外,铜外排转运蛋白 ATP7A 和 ATP7B 的上调导致卵巢癌产生化学耐药性。miR-139 可以直接与 ATP7A/B 的 3'-UTR 结合,从而诱导细胞凋亡并增加卵巢癌细胞的化学敏感性。
药物灭活
金属硫蛋白 (MT) 和谷胱甘肽 (GSH) 是两种主要的与铂类药物结合的含硫醇蛋白。细胞内含硫醇蛋白对顺铂的解毒作用被认为是需要克服的主要障碍。MT 与顺铂结合可诱导耐药性,而短发夹状 MT (shMT) 可以逆转这一现象。GSH 与顺铂反应形成 GS-铂复合物,从而降低细胞内可用的铂含量。谷胱甘肽 S-转移酶 (GST) 催化 GSH 与铂结合并导致药物失活,这与卵巢癌的铂耐药性有关 [ 36,37 ]。
DDR 中的改动
DNA损伤若得不到及时修复,细胞衰老或凋亡信号就会被激活,而DDR的异常激活则使癌细胞保持活力,显著诱导对化疗药物和PARPis的耐药性并影响治疗效果。DDR通常由七条通路组成(图 3):HRR、NHEJ、碱基切除修复(BER)、核苷酸切除修复(NER)、错配修复(MMR)、跨损伤DNA合成(TLS)和范康尼贫血(FA)通路。DNA损伤反应、DNA修复成分和miRNA之间存在相互作用已有基因解码。作为调控因子,miRNA的异位表达可干扰DNA修复机制的活性,而DNA修复机制与多种类型的耐药性有关。一些miRNA可以通过靶向编码DDR相关酶的基因来逆转耐药性。
图 3.DNA 损伤修复的改变
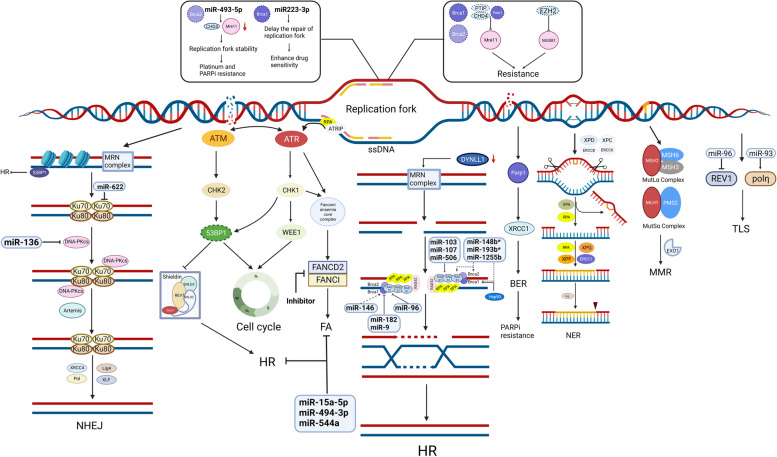
图3:DNA 损伤修复的改变。DDR 通常由 HRR、NHEJ、复制叉、BER、NER、MMR、TLS 和 FA 组成。DSB 的修复主要通过 NHEJ 修复途径与 HRR 途径结合进行。NHEJ 由 Ku70–Ku80 异二聚体与 DNA 末端结合而启动。随后 DNA-PKcs 的募集和自磷酸化将 DNA 末端聚集在一起,并允许它们通过 XRCC4–LIG4 连接。MRN 复合物 (MRE11-RAD50-NBS1) 是 HRR 的重要修复因子,它首先检测 DNA 损伤并激活下游信号传导。此外,它发挥核酸酶活性来切除 DNA 末端,从而引导 HRR。此外,DYNLL1 直接与 MRE11 结合以限制其末端切除活性。DYNLL1 减少可恢复 HR 介导的双链 DNA 断裂修复。复制叉保护是一种不依赖于DSB的机制,有助于基因稳定,从而导致化学抗性和PARPi抗性。此外,53BP1蛋白的下调是恢复DNA末端切除的另一种机制。Shieldin(SHLD1、SHLD2、SHLD3和REV7)作为53BP1的效应复合物,可以以BRCA非依赖的方式介导53BP1依赖的DNA修复。激酶ATR和ATM在DDR通路中起着至关重要的作用,例如维持复制叉稳定性和调节CHK1和CHK2。CHK1可以激活G2/M抑制激酶WEE1来维持基因组完整性。一些miRNA被证明可以调节参与HRR、NHEJ、复制叉保护、TLS和FA的成分的表达,但miRNA与BER/NER/MMR之间的相互作用缺乏足够的证据。 (SLC,溶质载体超家族;GST,谷胱甘肽转移酶;MT,金属硫蛋白)
同源重组修复
HR 缺乏是许多 HGSOC 病例(约 50%)的特征,被认为是对铂类药物和 PARPis 敏感性的预测生物标志物。HR 通路活性的恢复可能导致 HR 缺乏的卵巢癌患者对铂类药物和 PARPis 产生获得性耐药性。值得注意的是,已发现 miRNA 通过直接靶向 DDR 反应的成分来阻碍 DDR,从而降低耐药性。miR-146 靶向 BRCA1 并与对双链断裂 (DSB) 的反应相关。过表达的 miR-182 和 miR-9 介导 BRCA1 的下调并增加卵巢癌对顺铂和 PARPis 的敏感性 [ 46,47 ] 。 miR-96 直接靶向 RAD51 的编码区和 REV1 的 3'-UTR,并降低 HRR 的效率[ 43 ]。miR-1255b、miR-193b* 和 miR-148b*(“*”表示低浓度下的微量产物)可以分别靶向 HR 介导的 DSB 修复因子 BRCA1、BRCA2 和 RAD51 的转录本,从而调节 PARPi 敏感性[ 48 ]。miR-506、miR-103 和 miR-107 是卵巢癌患者化疗反应和生存的有力临床标志物,可以通过直接靶向 RAD51 和抑制 RAD51 病灶的形成来使癌细胞对 DNA 损伤敏感[ 49 , 50 ]。重要的是,在长期暴露于铂类药物和PARPi期间以及在进展后活检中发现了BRCA1 /2、RAD51C和PALB的回复突变。这些突变恢复了开放阅读框,从而恢复了HRR的功能[ 51,52 ]。此外,还发现HSP90介导BRCA1的稳定,BRCA1与PALB2-BRCA2-RAD51复合物相互作用。这种相互作用对于RAD51病灶的形成以及赋予PARPi和顺铂耐药性至关重要[ 53 ]。HSP90抑制剂和铂类的联合治疗是一种创新的抗肿瘤策略,有可能逆转卵巢癌的铂类耐药性[ 54,55 ]。
MRE11-RAD50-NBS1(MRN)复合物是 HRR 的一个重要因子,它首先检测 DNA 损伤,然后激活信号分子 [ 56 ]。此外,它还发挥核酸酶活性来切除 DNA 末端,从而引导 HRR。此外,重组人细胞质动力蛋白轻链 1(DYNLL1)被发现可直接与 MRE11 结合以限制其末端切除活性。因此,DYNLL1的下调可恢复 HR 介导的 DNA DSB 修复,从而诱导卵巢癌的化学耐药性和 PARPi 耐药性 [ 57 ]。此外, TP53BP1基因的功能丧失突变导致 53BP1 蛋白表达下降并促进 BRCA1 独立的 DNA 末端切除,这导致铂类和 PARPi 耐药性 [ 58 ]。
鉴于免疫检查点抑制剂作为治疗药物的作用不断扩大,肿瘤 DNA 损伤和修复与免疫反应的相互作用最近成为关注的焦点。研究发现,患有 BRCA 突变和同源重组缺陷 (HRD) 的 HGSOC 患者的 CD3 + /CD8 + 肿瘤浸润淋巴细胞 (TIL)、PD-1/PD-L1 免疫组织化学染色和新抗原载量增加。此外,患有 RAD51、ATM 和 ATR 突变的野生型 BRCA1/2 卵巢肿瘤的预测新抗原水平高于 HR 功能正常的肿瘤[ 59,60 ] 。Mu Chen 等人的研究表明,DNA 损伤导致细胞质中产生许多 DNA 片段,从而导致细胞表面抗原呈递增加并激活免疫反应[ 61 ]。然而,一项临床试验表明,阿维单抗并未显示其对BRCA1/2突变卵巢癌患者的疗效有所改善(NCT01772004)。因此,有必要进行更多临床试验,以确定 DNA 损伤和免疫调节剂之间相互作用的复杂性。
新近进展
NHEJ 通过在修复过程中与 HRR 竞争来修复 DNA DSB,其机制包括 TP53BP1、DNA-PK 等。[ 62 ] miRNA 在调节这些 NHEJ 相关基因的表达中起重要作用[ 39 ]。miR-136 过表达会下调 DNA-PK、细胞周期相关基因和抗凋亡基因,使卵巢癌细胞对紫杉醇重新敏感[ 63 ]。miR-622 通过靶向 Ku 复合物来抑制 NHEJ 并促进 HR 介导的 DSB 修复。因此,在 BRCA1 缺陷的 HGSOC 细胞中高表达的 miR-622 会诱导铂类和 PARPi 耐药性[ 64 ]。DNA-PK 由 DNA-PKcs 和 DNA 末端结合 Ku70/80 异二聚体组成,已成为 NHEJ 通路中一个有趣的治疗靶点[ 65,66 ] 。该异二聚体可以识别 DSB 并形成 Ku-DNA 复合物,后者可以募集 DNA-PK 到 DSB 位点[ 67 ]。DNA-PKcs 通过自身磷酸化和募集下游效应物(如内切酶(Artemis)[ 68 ]和聚合酶(DNA POLM(Pol µ)和 POLL(Pol λ))[ 69 , 70 ])在促进 NHEJ 中起主要作用。研究发现,DNA-PK 抑制可诱导 HR 功能恢复,并导致患者来源的卵巢癌异种移植对 PARPis 产生抗性[ 71 ]。XRCC5/Ku80 [ 66 ] 和 XRCC6/Ku70 [ 65 ] 的异位表达会诱导铂类和 PARPi 抗性。至关重要的是,TP53BP1 可以通过限制 DSB 切除和拮抗 BRCA1 缺陷细胞中的 BRCA2/RAD51 加载来促进 NHEJ 并减少 BRCA1 介导的 HRR [ 72 ]。Shieldin 复合物(由 SHLD1、SHLD2 和 SHLD3 组成)是 53BP1 的效应复合物,可在各种环境下调节 53BP1 依赖性 NHEJ,并影响 HRD 缺陷细胞对 PARPis 的抵抗力 [ 73 , 74 ]。最后,XRCC4、DNA 连接酶 IV (LIG4) 和 XLF 是末端连接的核心成分。
复制叉保护
复制叉保护以独立于 DSB 诱导的 HR 的方式促进基因组稳定性,从而导致化学抗性和 PARPi 抗性[ 75 ]。PARP1、BRCA1 和 BRCA2 在复制压力 (RS) 条件下在保护复制叉方面起关键作用[ 76 , 77 ]。BRCA 缺陷细胞中的 PTIP、PARP1 和 CHD4 缺陷会阻止 MRE11 核酸酶的募集以阻止复制叉,进而保护新生 DNA 免于降解,从而产生化学抗性和 PARPi 抗性[ 78 ]。在 BRCA2 突变的细胞和患者中,EZH2 下调导致 MUS81 核酸酶被抑制,从而恢复 DNA 复制叉保护,导致 PARPi 抗性[ 79 ]。 miRNA-493-5p 通过下调MRE11和CHD4显著维持 BRCA2 突变卵巢癌细胞中的复制叉稳定性,从而产生铂和 PARPi 耐药性 [ 10 ]。然而,恢复 miR223-3p 表达会延迟复制叉的修复,导致基因组不稳定,并增强 BRCA1 缺陷型 OC 的药物敏感性 [ 80 ]。
NER 和 BER
NER 负责修复单链 DNA 损伤,根据癌症基因组图谱 (TCGA) 数据库,8% 的 HGSOC 患者表现出某些 NER 基因的改变[ 81 ]。NER 信号通路可以修复铂诱导的加合物,因此,NER 基因的上调,包括 ERCC1、ERCC2-XPD、ERCC3-XPB、ERCC4-XPF、ERCC5-XPG、ERCC6、ERCC8 和 XPA,可能介导化学耐药性[ 63 ]。事实上,ERCC1 或 XPF 的过表达不仅增加了铂类耐药性,而且降低了奥拉帕尼的毒性[ 82 ]。虽然某些 NER 基因突变(ERCC6-Q524* 和 ERCC4-A583T)在体外发现与铂类敏感性功能相关,但这些 NER 改变并不影响 HR 或赋予对 PARPis 的敏感性。
PARPs 和支架蛋白 XRCC1 加速了 BER。目前有报道称 BER 通路与铂类耐药性既有正相关性,也有负相关性。虽然 BER 通路中间体是 PARPis 疗效的基础,但它们介导 PARP 家族蛋白(尤其是 PARP1)的活性以启动修复,从而导致 PARPi 耐药性。
MMR 缺陷
尽管 OC 中的 MMR 缺陷是继 BRCA1/2 突变之后导致遗传性卵巢癌的最常见原因,但对其的研究相对较少。MMR 通路包含七种蛋白质(MSH2、MSH3、MSH6、MLH1、MLH3、PMS1 和 PMS2)[ 66 ]。据报道,卵巢癌患者中 MMR 缺陷(任何蛋白质缺失)的发生率为 2% 至 29% [ 67 ]。少数研究表明 MMR 缺陷与耐药性有关,但结果尚无定论 [ 83 – 86 ]。MMR 缺陷在卵巢癌耐药性中可能发挥的作用值得进一步研究。目前,MMR缺陷被认为是由于无效MMR活性的丧失,复制叉停滞,无法识别DNA损伤,顺铂加合物的净复制旁路增加以及重组依赖性旁路水平的调节而发生的[ 87,88 ] 。
其他 DDR 途径
FA核心复合物由至少10种FA相关蛋白组成(FANCA、FANCB、FANCC、FANCE、FANCF、FANCG、FANCL、FAAP100、FAAP20 和 FAAP24)[ 89 ]。抑制FA修复途径的成分,如FA互补组D2(FANCD2)和FANCI,可增加对化疗药物的敏感性[ 90 ]。miR-15a-5p、miR-494-3p和miR-544a可能抑制整个FA/HR通路[ 91 ]。
TLS 由 DNA 聚合酶(例如 Pol η 和 REV1)介导。它增加肿瘤细胞对铂诱导的 DNA 加合物的耐受性,并导致铂耐药性 [ 92 ]。Pol η 和 REV1 是跨损伤 DNA 聚合酶 [ 93 ]。miR-93 的上调可能通过靶向 DNA Pol η 来逆转耐药性 [ 92 ]。据报道,miR-96 可通过抑制 REV1 介导的 TLS 来防止化学耐药性的出现。
失调的癌症相关信号通路
一系列信号通路(图 4)共同调节人类恶性肿瘤中的生物过程,并与癌细胞的增殖、侵袭和治疗耐药性有关 [ 94 ]。miRNA 可通过 miRNA-mRNA 结合来调节信号通路成分的表达,通常是结合到 mRNA 3'-UTR 中的 miRNA 靶位 [ 40 , 95 ]。尽管与癌症相关的信号通路很复杂,但确定潜在的治疗靶点仍是大有可为的。
图 4.
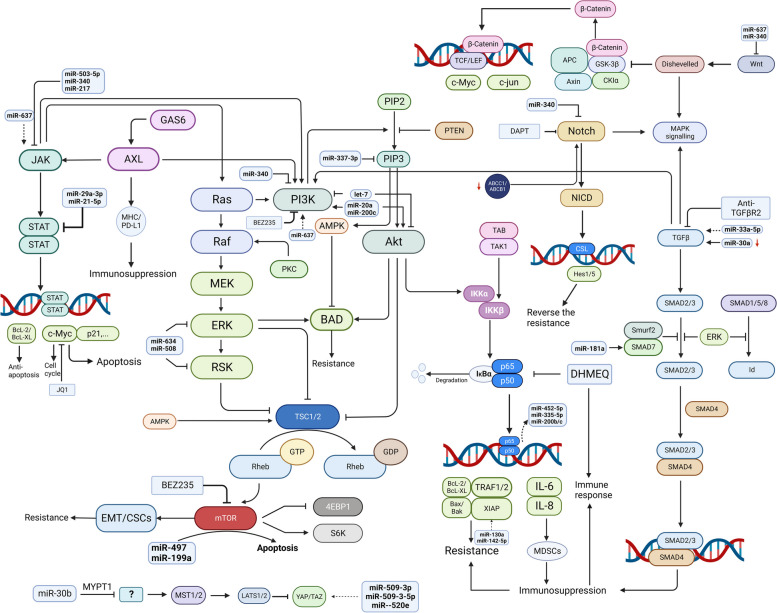
在新标签页中打开
肿瘤相关信号通路失调。一系列信号通路共同调控人类恶性肿瘤的生物学过程,与肿瘤的增殖、侵袭和治疗耐药性有关。信号通路主要包括NFκB、PI3K/Akt、JAK/STAT、Notch、GAS6/AXL、TGF-β、MAPK、Hippo/YAP通路。一些miRNA能够调控上述通路的关键成员,包括JAK/STAT、GAS/AXL、MAPK、PI3K/Akt、NFκB、TGF-β、Hippo/YAP,但尚无关于卵巢癌中miRNA与Notch相互作用的研究。失调的肿瘤相关信号通路干扰细胞凋亡、细胞周期和免疫状态,导致多药耐药。这些通路中的分子靶点可能为卵巢癌的耐药性提供新的途径。 γ-分泌酶抑制剂DAPT,c-Myc靶向小分子JQ1,NFκB DHMEQ的抑制剂,抑制OC增殖,诱导细胞凋亡,逆转药物耐药性。(JQ1,新型细胞通透性小分子;BAD,Bcl-2死亡拮抗剂;IKKα,核因子-κB亚基-α的抑制剂;mTOR,雷帕霉素的哺乳动物靶点;NF-κB,核因子-κB;DHMEQ,脱羟甲基环氧喹诺星;MDSC,髓系抑制细胞;CSC,癌症干细胞;BEZ235,双重PI3K / mTOR抑制剂;DAPT,γ-分泌酶抑制剂N-[N-(3,5-二氟苯乙酰基)-L-丙氨酰]-S-苯基甘氨酸叔丁酯)
NFκB信号通路
NFκB 在卵巢癌中可发挥双相功能。它在卵巢癌细胞中起抗癌作用,使它们对卡铂和紫杉醇诱导的细胞凋亡敏感,但它也具有致癌作用,可促进卵巢癌细胞的侵袭性和化学耐药性,并导致对这些治疗药物的耐药性[ 96 ]。常见的化疗药物,包括紫杉烷类、铂类药物、长春花生物碱和厄洛替尼,可激活 NFκB 及其促存活下游靶点,从而导致化学耐药性[ 97 ]。NFκB 通路的激活与铂类耐药性相关,并导致卵巢癌患者预后不良[ 98 ]。从机制上看,p65 亚基核转位增加和 IκB 激酶抑制剂亚基 α 和 β 的磷酸化是 NFκB 活化的标志,可促进化疗耐药性 [ 99 ]。此外,NF-κB p65 通过与 miR-200b/c 启动子结合来增加 miR-200b/c 的表达,从而提高卵巢癌细胞对顺铂的敏感性 [ 100 ]。它还通过 NF-κB TFs RelA 和 RelB 调节下游 miRNA miR-452-5p 和 miR-335-5p,防止卵巢癌复发 [ 101 ]。此外,NF-κB信号通路与卵巢癌细胞的免疫抑制和免疫逃逸有关,部分是通过NFκB依赖性IL-6的产生来实现的,IL-6会损害树突状细胞(DC),但会生成和募集免疫抑制性MDSC,而IL-8则会增强免疫抑制酶精氨酸酶的表达[ 102 ]。脱羟甲基环氧喹诺星(DHMEQ)是一种NFκB抑制剂,可诱导细胞凋亡,增强对铂类药物的应答,并逆转卵巢癌细胞的免疫抑制[ 102,103 ] 。
PI3K/Akt 通路
PI3K/Akt 通路在卵巢癌中经常上调,激活的 PI3K/Akt 信号可增强癌细胞化学耐药性[ 104,105 ]。已发现许多 miRNA 可调节 PI3K/Akt 通路,影响卵巢癌化学敏感性[ 106 ]。miR-337-3p 直接靶向 PIK3CA 和 PIK3CB,抑制上皮性卵巢癌细胞增殖并逆转耐药性[ 107 ]。let - 7 miRNA 家族通过控制 PI3K 和 Akt1 磷酸化和活性来失调该通路[ 108 ]。然而,miR-20a 和 miR-200c 激活并上调该通路,导致紫杉醇耐药性[ 109 ]。肿瘤细胞中异常的 PI3K-Akt 信号转导归因于铂类耐药表型,而顺铂与 LY-294002(一种 PI3K-Akt 双激酶抑制剂)联合使用被发现可阻止 3D 球体形成并使细胞对顺铂敏感[ 110 ]。此外,mTOR 是 PI3K/Akt 通路中关键的下游信号激酶[ 111 ]。激活的 mTOR 信号转导可触发上皮间质转化 (EMT) 并促进癌症干细胞 (CSC) 的维持,从而导致卵巢癌患者产生化学耐药性,而使用 BEZ235(一种双重 PI3K/mTOR 抑制剂)治疗可能是逆转化学耐药性的一种有前途的方法[ 112 ]。此外,研究发现,miR-497 和 miR-199a 可以定量控制 mTOR 表达,从而诱导卵巢癌细胞凋亡[ 106 ]。
JAK/STAT通路
JAK 磷酸化后,STAT 也被磷酸化并激活,随后其核易位诱导参与生长和凋亡的靶基因转录。M Koti 等人报道,STAT1 是化学耐药和化学敏感性 HGSOC 之间最显著的差异表达基因。STAT1 的上调与铂类耐药性有关 [ 113 ]。c-Myc 是 JAK/STAT 信号通路的下游靶点,与 OC 的恶性肿瘤和化疗反应有关 [ 114 ]。新型细胞通透性小分子 JQ1 可以靶向 c-Myc 来抑制 OC 细胞增殖并诱导其凋亡。与化疗药物和 PARPis 一样,JQ1 值得进一步研究,以了解其通过与 JAK-STAT 信号通路相互作用逆转 OC 患者耐药性的能力 [ 115 ]。该通路也受 miRNA 调控,miRNA 相互作用与耐药性有关。恢复 miR-503-5p 表达可通过将该 miRNA 与介质 CD97 的 3’-UTR 结合来阻断下游 JAK2/STAT3 通路[ 116 ]。miR-340 还可以直接靶向 LGR5、FHL2、CTNNB1 和 BAG3,分别抑制 JAK/STAT3、Wnt/β-catenin、Notch 和 PI3K/Akt 通路[ 117 ]。miR-637 受竞争性内源性 RNA (ceRNA) 调控,并参与 OC 中的五种信号通路,包括 JAK/STAT3、Wnt/β-catenin 和 PI3K/Akt 信号通路[ 118 ]。此外,JAK/STAT 通路可以通过塑造免疫细胞浸润对卵巢癌发挥作用。干扰素介导的 STAT1 激活导致下游靶标 CXCL10 的表达,而 CXCL10 是效应 Th1 CD4+ 细胞、自然杀伤 (NK) 细胞和 CD8+ 细胞运输和分化的关键[ 113 ]。此外,miR-217 过表达介导的 JAK/STAT3 信号通路减弱可抑制 M2 巨噬细胞极化并调节免疫状态[ 119 ]。
Notch信号通路
Notch 信号通路由配体与 Notch 受体结合激活。γ-分泌酶(Notch 通路中的一种重要蛋白水解酶)对 Notch 进行蛋白水解切割后,活性 NICD 片段易位到细胞核中,并通过与 CSL 转录调节因子相互作用诱导 Notch 靶基因转录 [ 120 ]。异常的 Notch 通路可导致卵巢癌细胞产生耐药性,而 Notch 敲低可通过下调 ABCC1 和 ABCB1 增加铂类敏感性 [ 121,122 ]。此外,抑制 Notch 信号通路可诱导细胞凋亡并逆转耐药性。 γ-分泌酶抑制剂N-[N-(3,5-二氟苯乙酰基)-L-丙氨酰]-S-苯基甘氨酸叔丁酯 (DAPT) 可通过下调 Notch 信号诱导细胞凋亡,进而逆转卵巢癌细胞的铂类耐药性 [ 123,124 ]。此外,抑制 Notch 信号可增加动物模型中卵巢癌细胞的凋亡,并逆转对顺铂和紫杉醇的耐药性[ 121,125 ]。
GAS6/AXL 通路
GAS6 与 AXL 结合导致 AXL 二聚化和酪氨酸残基处的自身磷酸化,从而导致细胞内信号转导[ 126 ]。GAS6/AXL 通路通过与其他信号相互作用和调节肿瘤微环境 (TME) 来影响耐药性。例如,AXL 相关的 EMT 介导对化疗和靶向治疗的耐药性[ 127,128 ]。在卵巢癌中,GAS6/AXL 通路还通过与其他信号通路(如 PI3K、JAK/STAT 和 MAPK 通路)相互作用产生耐药性[ 129 ]。此外,GAS/AXL 通路在卵巢癌 DDR 中的作用已逐渐显现。抑制 AXL(通过 bemcentinib 或 MYD1-72)可通过增加 DNA 损伤和诱导 RS使卵巢癌细胞对铂、ATR 抑制剂 (ATRis) 和 PARPis 重新敏感[ 130-132 ]。此外,GAS6/AXL 信号通过调节肿瘤细胞中 MHC 和 PD-L1 的表达、增加免疫抑制趋化因子的分泌以及干扰免疫细胞的浸润来促进免疫抑制性 TME 的产生[ 133 ]。尽管 miR-515-3p 部分通过靶向 AXL 来调节粘液性卵巢癌中的奥沙利铂敏感性[ 134 ],但仍然缺乏足够的证据证明 miRNA 在调节 GAS6/AXL 通路中的作用。
转化生长因子-β (TGF-β) 通路
TGF-β 信号通路的激活是通过二聚 TGF-β 配体与其特异性跨膜受体的相互作用而发生的[ 135 ]。TGF-β 信号通过下游 SMAD 效应子和非 SMAD 蛋白(如 AKT 和 MAPK)进行转导[ 136 ]。miRNA 可靶向 TGF-β 信号通路的成分,介导卵巢癌的耐药性。例如,miR-33a-5p 通过靶向肉碱 O-辛酰转移酶 (CROT) 影响 SMAD2/4 的表达,从而诱导卵巢癌对紫杉醇的耐药性[ 137 ]。miR-30a 表达降低可导致 TGF-β 和 SMAD4 上调,最终激活自噬,介导卵巢癌对顺铂的耐药性[ 138 ]。然而,miR-181a 通过直接靶向 SMAD7 激活 TGF-β 信号传导,在介导 HGSOC 耐药性方面发挥着未被重视的作用[ 139 ]。
TGF-β 通路具有双相作用,在早期起到肿瘤抑制因子的作用,但后期通过影响肿瘤细胞及其微环境刺激癌症进展[ 135 ]。该通路的异常激活会阻止细胞凋亡并导致卵巢癌细胞产生化学耐药性[ 140 ]。此外,TGF-β 通路通过典型的下游 EMT 相关分子在铂类耐药性中起着至关重要的作用[ 141 ]。TGF-β 通路还抑制 TME 内的免疫并导致化学耐药性。Daniel Newsted 等人开发了一种抑制抗体 (抗 TGFBR2) 来阻断 TGF-β 信号传导,并表明这种抗体提高了化疗的疗效和有限的抗肿瘤免疫反应[ 142 ]。此外,可通过 CRISPR/Cas9 介导的 TIL 中 TGF-β 受体 2 (TGFBR2) 敲除来诱导 TGF-β 信号通路的免疫抑制作用[ 143 ]。
MAPK 通路
RAS/RAF/MEK/ERK 是 MAPK 通路中经典且关键的信号传导介质,卵巢和腹膜低级别浆液性癌 (LGSC) 以 MAPK 通路改变和化学耐药性为特征[ 144 ]。Ras 和 Erk1/2 的过度活化与卵巢癌的化学耐药性呈显著正相关[ 145 ]。PI3K/Akt 和 Ras/MAPK 信号通路均可介导促凋亡蛋白 BAD 的磷酸化,从而通过抑制细胞凋亡导致铂类耐药性增加[ 146 ]。miRNA 也通过干扰 MAPK 通路的成分发挥调节作用。例如,miR-634 可以直接抑制 GRB2、ERK2 和 RSK2,因此,抑制 Ras-MAPK 通路可恢复卵巢癌细胞的化学敏感性[ 147 ]。在卵巢癌中发现了低水平的 miR-508/miR-18a 和增加的 MAPK1 和 ERK 表达,而发现 miR-508 模拟物抑制 MAPK1 和 ERK,从而抑制 EMT 和癌细胞的恶性进展[ 148,149 ]。
Hippo/yes 相关蛋白 (YAP) 通路
Hippo 通路导致卵巢癌常用于治疗药物的耐药性 [ 150 , 151 ]。YAP 及其同源物 TAZ 是 Hippo-YAP 通路的主要下游效应物,并充当转录辅激活因子,它们的信号转导已成为药物耐药性的关键机制 [ 152 , 153 ]。YAP 和 TAZ 通过结合 TF(如 TEA 结构域家族 (TEAD) 蛋白)介导基因转录,从而促进肿瘤进展和耐药性 [ 153 , 154 ]。miRNA 可以调节 YAP1 的表达并调节 Hippo 通路,但所涉及的调控机制仍不清楚。miR-509-3p、miR-509-3-5p [ 155 ] 和 miR-141 [ 156 ] 通过 YAP1 和 Hippo 信号通路与顺铂耐药性相关。有假设称,miR-509-3-5p 可以通过靶向其编码区来直接调控 YAP1 表达[ 155 ]。
表观遗传修饰
表观遗传调控是指在DNA序列未发生改变的情况下,基因表达发生可遗传的变化。DNA甲基化、组蛋白修饰和非编码RNA(ncRNA)活性(图 5)是常见的表观遗传调控机制[ 157 ]。越来越多的证据表明,表观遗传调控异常可导致肿瘤耐药。
图 5.
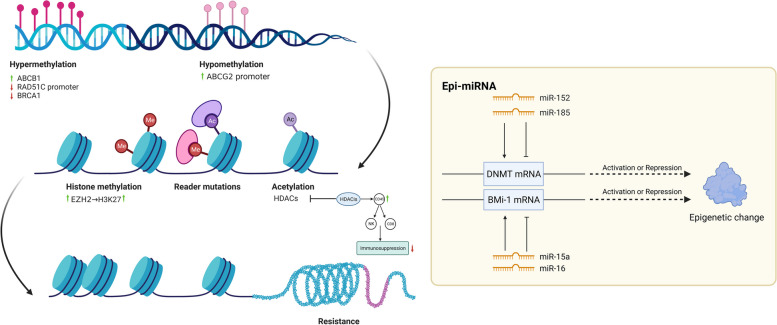
在新标签页中打开
表观遗传修饰。表观遗传过程通过 DNA 甲基化、组蛋白修饰和非编码 RNA (ncRNA) 来调节基因表达,而无需改变 DNA 序列。ABCB1 的高甲基化和 ABCG2 启动子的去甲基化导致卵巢癌产生化学耐药性。已证实 RAD51C 启动子甲基化的丧失和 BRCA1 甲基化的下调会导致耐药性。特异性 H3K27 甲基转移酶 EZH2 通过 H3K27 甲基化赋予卵巢癌细胞化学耐药性。miRNA 的一个子类“epi-miRNA”可以调节表观遗传调节因子以影响治疗反应。miR-152 和 miR-185 通过直接靶向 DNMT1 共同导致顺铂耐药性,miR-15a 和 miR-16 直接靶向 Bmi-1(Polycomb 复合物的成员)。它们可能成为潜在的表观遗传治疗靶点。包括DNMTi和HDACi在内的表观遗传治疗可以增加TME中CD45+免疫细胞、活性CD8+T细胞和NK细胞的数量,降低免疫抑制。因此,表观遗传治疗联合免疫治疗可能是治疗耐药性OC的一种有前途的治疗策略。(HDACs,组蛋白去乙酰化酶;H3K27,组蛋白H3赖氨酸27;EZH2,zeste同源物2增强子;DNMTis,DNA甲基转移酶抑制剂;HDACis,组蛋白去乙酰化酶抑制剂;Bmi-1:Polycomb复合物成员)
DNA甲基化可通过多种机制影响治疗反应,包括影响膜转运、DNA修复、信号通路活性和细胞凋亡[ 158 ]。例如,ABCB1高甲基化和ABCG2启动子去甲基化可能影响治疗效果并导致卵巢癌产生化学耐药性,这种影响归因于P-gp的上调[ 159,160 ] 。PI3K-AKT、MAPK和Wnt通路以及EMT相关基因的异常甲基化使HGSOC细胞产生耐药性[ 161-163 ]。此外,RAD51C启动子甲基化的缺失和BRCA1甲基化的低水平已被证实会导致耐药性。纯合的RAD51C甲基化和BRCA1高甲基化可以作为HGSOC治疗反应的预测生物标志物[ 164 ]。对接蛋白 2(DOK2)基因的表观遗传改变可通过抑制细胞凋亡诱导卵巢癌产生卡铂耐药性[ 165 ]。
组蛋白修饰主要包括组蛋白甲基化和乙酰化[ 166 ]。Min-Gyun Kim等证实了卵巢癌细胞系SKOV3和OVCAR3中组蛋白去乙酰化酶(HDAC)过表达与顺铂耐药之间的相关性[ 167 ]。最近的数据为组蛋白H3赖氨酸27(H3K27)甲基化在耐药机制中的作用提供了新的见解[ 168 ]。zeste同源物2(EZH2)的特异性H3K27甲基转移酶增强子通过H3K27甲基化赋予卵巢癌细胞化学耐药性[ 169 ]。此外,Yujie Fang等。基于对 TCGA 和基因表达综合 (GEO) 数据库的分析,揭示了组蛋白乙酰化在卵巢癌弱免疫反应和化学耐药中的作用[ 170 ]。在治疗方面,表观遗传治疗,包括用 DNA 甲基转移酶和组蛋白去乙酰化酶抑制剂(分别为 DNMTis 和 HDACis)治疗,可以增加 TME 中的 CD45 + 免疫细胞、活性 CD8 + T 细胞和 NK 细胞的数量,通过激活小鼠卵巢癌中的 I 型干扰素信号传导来减轻免疫抑制和肿瘤负担[ 171,172 ]。
NcRNA 包括长 NcRNA (lncRNA)、小 NcRNA (sncRNA) 和环状 RNA (circRNA),可通过表观遗传修饰调节基因表达 [ 173 ]。最常见的是,lncRNA 和 circRNA 通过充当 miRNA 海绵来调节下游基因表达,在耐药性中发挥作用 [ 174 ]。“表观 miRNA”通过直接靶向表观遗传调节因子发挥作用,如 DNMT 和 HDAC,或多梳阻遏物复合物的成分[ 175 ]。miRNA 通过结合与 mRNA 3'-UTR 结合来影响 mRNA 转录,从而恢复高甲基化的肿瘤抑制基因的表达[ 176 ]。下调的 miR-152 和 miR-185 通过直接靶向 DNMT1 共同导致顺铂耐药性,因此可作为表观遗传治疗靶点 [ 177 ]。 miR-15a 和 miR-16 直接靶向 Bmi-1(Polycomb 复合物的组成部分)的 3’-UTR,其表达水平与卵巢癌中的 Bmi-1 蛋白水平显著相关[ 178 ]。
其他机制
事实上,确定卵巢癌复杂的耐药机制仍然极具挑战性。耐药机制相互交叉,可能通过产生免疫抑制环境而互相干扰,从而导致耐药性,包括免疫疗法耐药性。Treg/Th17 细胞失衡[ 179 ]、巨噬细胞 M2 极化[ 180 ]、NK 细胞耗竭[ 181 ]以及 IFNγ[ 182 ]和 PD-L1[ 183,184 ]的异常表达会介导免疫抑制,促进肿瘤进展和耐药性。miRNA(如 miR-29a-3p、miR-21-5p、 miR -1246、miR-29c 和 miR-424)可以调节免疫相关分子的表达,从而影响免疫状态。相反,TME 或免疫疗法可以调节许多 miRNA 的表达,从而促进耐药性 [ 185 , 186 ]。Hedgehog (Hh) 和 Wnt/β-catenin 通路也可以促进 T 细胞排斥和检查点抑制剂耐药性 [ 187 , 188 ]。然而,在一项 II 期临床试验 ( NCT00739661 )中,Hh 通路抑制剂 vismodegib 单药治疗在卵巢癌患者中未表现出任何显著的抗肿瘤活性[ 189 ]。有趣的是,尽管 Wnt 信号是卵巢癌耐药性的驱动因素,但 Wnt 信号的遗传驱动因素在很大程度上是未知的 [ 190 ]。
除了上述机制外,细胞凋亡、铁死亡、自噬和内质网应激 (ER 应激) 的异常同时或依次作用,使癌细胞能够在抗肿瘤药物治疗下存活。据报道,miR-130a [ 191 ] 和 miR-142-5p [ 192 ] 通过靶向 XIAP 来调节细胞凋亡。对铁死亡的深入研究表明,铁死亡在获得性索拉非尼耐药性 [ 193 ]、EGFR 酪氨酸激酶抑制剂耐药性 [ 194 ] 和免疫疗法耐受性 [ 195 ] 中起着关键作用。有趣的是,紫杉醇可驱动自噬通量,从而促进卵巢癌对紫杉醇产生耐药性[ 196 ],而 miR-30a[ 138 ]、miR-200c[ 197 ]和 miR-133a[ 198 ]可调控自噬通量。此外,作为一个热门的研究课题,内质网应激对卵巢癌的耐药性有相当大的影响[ 199 ]。内质网应激期间,IRE1α/XBP1s 通路可激活未折叠蛋白反应 (UPR),导致微环境重塑或对治疗产生耐药性[ 199 , 200 ]。
克服耐药性的策略
针对跨膜转运的临床试验
ABCB1 (也称为 p-gp/MDR1) 的过度表达会介导药物外排增加。药物外排增加使得达到足够的细胞内药物浓度变得困难,从而导致耐药性 [ 201,202 ] 。ABCB1 抑制剂 (维拉帕米和 elacridar) 可通过减少许多药物的外排来逆转 MDR,包括紫杉醇、奥拉帕尼、阿霉素和鲁卡帕尼 [ 24 ]。此外,在小鼠模型中评估了 PARPi 耐药性,发现通过共同使用 tariquidar (一种 P-gp 抑制剂) 可以逆转这种耐药性 [ 26 ]。尽管已经进行了对 P-gp 抑制剂反应的临床前研究,但由于这些药物的严重毒性作用,P-gp 抑制剂的临床试验有限且过时 [ 203 ]。例如,P-gp 抑制会增加紫杉醇在细胞内的积累,导致紫杉醇诱导的周围神经病变 [ 204 ]。ncRNA 在调节 ABC 转运蛋白及其对 MDR 的临床意义方面起着关键作用 [ 8 ]。因此,耐药后治疗的新策略包括递送 ncRNA 模拟物或 ncRNA 的反义寡核苷酸以干扰 ncRNA-ABC 转运蛋白轴。此外,通过多肽纳米颗粒共同递送 miR-129-5p 和阿霉素被发现可通过直接抑制 P-gp 来有效克服 MDR,从而增加细胞内阿霉素的积累并增强化学敏感性 [ 205 ]。
近来,抗体药物偶联物 (ADC) 受到越来越多的关注,它可以直接将强效的细胞毒药物递送至具有适当靶抗原的癌细胞,同时避免对健康细胞产生毒性作用。目前,唯一获得 FDA 批准的 ADC 即米尔维妥昔单抗索拉坦新,在卵巢癌耐药的背景下引起了广泛关注。一项 III 期临床试验 MIRASOL ( NCT04209855 ) 正在进行中,旨在比较化疗和米尔维妥昔单抗索拉坦新在 FRα 阳性、铂类耐药 HGSOC 中的疗效。新型 ADC BA3011 可以通过条件活性生物制剂技术靶向癌细胞上的 Axl 受体。一项 II 期临床试验正在进行中,以评估 BA3011 和 durvalumab 联合治疗铂类耐药 HGSOC 患者 ( NCT04918186 )。 MUC16 是铂类耐药性卵巢癌治疗的另一个常见靶点,已在两项已完成的 I 期试验(NCT01335958 [ 206 ] 和NCT02146313 [ 207 ])中进行了评估。结果表明,抗 MUC16 ADC 在 MUC16 高表达的铂类耐药性卵巢癌患者中具有可耐受的安全性和令人鼓舞的抗肿瘤活性。此外,一些 miRNA 的下调可能导致 OC 中 MUC16 水平异常。因此,对于 OC 患者,它们的上调或模拟物可以与抗 MUC16 一起成为潜在的选择[ 208 ]。间皮素是一种在癌细胞表面过度表达的糖蛋白。两项 I 期临床试验(NCT01469793 / NCT02751918)评估了一种新型抗间皮素 ADC 在铂类耐药性卵巢癌中的应用。从这些试验得出的结论表明,anetumab ravtansine 与聚乙二醇化脂质体阿霉素联合使用具有良好的耐受性和良好的临床活性 [ 209 ],尽管先前试验的结果并不一致。另一种 ADC 药物 Zilovertamab Vedotin 以 ROR1 为靶点,已在 II 期临床试验中应用(NCT04504916)。ROR1 也可以被 miR-382 靶向,这可能成为 OC 的另一种选择 [ 210 ]。此外,HER2、TROP2、DLL3 和 Nectin-4 是 ADC 的主要靶点。在进一步的研究和临床试验中,与 ADC 的联合策略已显示出作为新兴疗法的巨大潜力 [ 211 ]。
针对 DDR 的临床试验
HRR 通路是卵巢癌获得性铂和 PARPi 耐药性的关键机制之一。DNA 修复靶向治疗是一种有前途的卵巢癌精准医疗策略。许多临床试验,包括评估针对 ATR、ATM、WEE1、检查点激酶 1/2 (CHK1/2)、BRCA1/2 和 RAD51 的药物的试验,都旨在评估干扰 DDR 通路以克服卵巢癌的铂和 PARPi 耐药性。
ATR/ATM 激酶抑制剂
ATR/ATM 激酶是 DDR 中的关键分子,是克服卵巢癌耐药性的潜在治疗靶点。据报道,miR-203a-3p 模拟物和 ATMis 可协同阻碍 OC 进展,这可作为 OC 的潜在治疗选择 [ 212 ]。据报道,ATRi 可以通过阻断 RAD51 加载到 DSB 上并破坏人源细胞系中的分叉保护来逆转 PARPi 耐药性 [ 213 ]。越来越多的临床试验评估了 ATRi 或 ATMi 与化疗药物或 PARPis 联合使用的疗效。一项介入交叉 II 期随机临床试验 ( NCT02595892 ) 是 ATRi 的首个随机临床试验,证明了在吉西他滨中添加 berzosertib 对治疗铂类耐药 HGSOC 的益处 [ 214 ]。 M4344 可增强临床 DNA 损伤药物(包括拓扑异构酶抑制剂、吉西他滨、顺铂和他拉帕尼)在晚期实体瘤中的活性[ 215 ]。最近,另一项针对 PARPi 耐药 HGSOC 患者的单组介入性 I 期试验 ( NCT04149145 ) 刚刚公布,其中将评估 M4344(一种 ATRi)与尼拉帕尼的联合方案。
WEE1 抑制剂
WEE1 是 HRR 通路中的重要靶点,许多正在进行的临床试验广泛评估了 WEE1 抑制剂与化疗药物或 PARPis 的联合使用。一项针对铂类耐药卵巢癌患者的 Ib 期非随机多中心研究 ( NCT04516447 ) 评估了 ZN-c3 与卡铂、PLD、紫杉醇和吉西他滨分别联合使用的临床前活性。两项 II 期试验 ( NCT01164995和NCT02272790 ) 测试了 WEE1 抑制剂 MK-1775 与卡铂或盐酸吉西他滨的联合使用。Adavosertib 联合化疗在铂类耐药卵巢癌中显示出初步治疗效果,但这种联合使用的血液学毒性可能会限制其应用[ 216,217 ] 。此外,WEE1 抑制剂 ZN-c3 与尼拉帕尼联合治疗铂类耐药卵巢癌患者的 I/II 期临床试验已开展(NCT05198804),但尚未公布结果。此外,会议摘要(ASCO 2021)报告称,adavosertib 单独使用或与奥拉帕尼联合使用对 PARPi 耐药患者均有疗效。虽然 3 级和 4 级毒性可以控制,但它们导致剂量中断和减少(NCT03579316)。
CHK1/2 抑制剂