
引言
亨廷顿疾病(Huntington’s disease, HD)是一种遗传性神经退行性疾病,由单个基因Huntingtin(HTT)缺陷导致,其特征是运动控制丧失以及认知能力和情绪稳定性的下降,常表现为不自主的舞蹈动作,因此又名亨廷顿舞蹈病【1】。该病遗传方式是显性遗传,HTT基因的一个特定区域(CAG重复区)会异常扩展,该片段编码一个长度可变的ployQ链。当这个polyQ链长度超过35个重复时,会导致蛋白质功能异常,进而损害脑细胞【2】。更长的polyQ链倾向于形成寡聚体(oligomers)、聚集体(aggregates)和包涵体(inclusion bodies),这些结构的形成被认为是导致神经毒性的源头【3】。关于HD发病机制还有另外一种假说,CAG重复序列可能通过RNA层面的毒性作用影响疾病的发展【4】。虽然已知突变型mHTT蛋白会产生聚集,但聚集与神经毒性之间的联系仍不清楚。
近日,来自美国斯坦福大学的Judith Frydman课题组在Nature Cell Biology上发表了研究论文Polyglutamine-mediated ribotoxicity disrupts proteostasis and stress responses in Huntington’s disease。在本研究中,作者探讨了HTT蛋白翻译和聚集的调控机制,发现了CAG扩展的突变体HTT蛋白通过核糖体毒性导致翻译过程中的过早终止,从而释放出易于聚集的mHTT蛋白,并进一步破坏细胞内蛋白质稳态和应激功能。这些发现不仅加深了科学家对HD发病机制的理解,也为其治疗提供了潜在的干预方案。

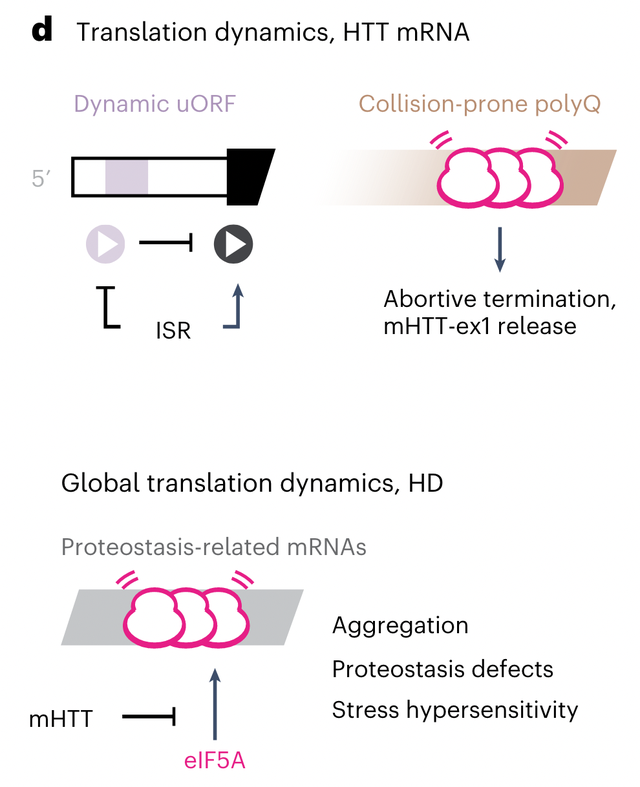 模式图(Credit: Nature Cell Biology)总的来说,这篇文章发现了降低mHTT水平和对抗其细胞毒性的靶点,CAG扩张会因核糖体碰撞而导致翻译终止,释放出具有神经毒性的mHTT片段,从而通过聚集隔绝eIF5A和其他蛋白质质量控制因子,导致神经元细胞蛋白质稳态障碍,抑制保护性应激反应的诱导。这些发现将有助于指导HD疾病治疗,提供了全新的干预策略。参考文献
模式图(Credit: Nature Cell Biology)总的来说,这篇文章发现了降低mHTT水平和对抗其细胞毒性的靶点,CAG扩张会因核糖体碰撞而导致翻译终止,释放出具有神经毒性的mHTT片段,从而通过聚集隔绝eIF5A和其他蛋白质质量控制因子,导致神经元细胞蛋白质稳态障碍,抑制保护性应激反应的诱导。这些发现将有助于指导HD疾病治疗,提供了全新的干预策略。参考文献1. Bates, G. P. et al. Huntington disease. Nat. Rev. Dis. Prim. 1, 15005 (2015).
2. Finkbeiner, S. Huntington’s disease. Cold Spring Harb. Perspect. Biol. 3, a007476 (2011).
3. Jimenez-Sanchez, M., Licitra, F., Underwood, B. R. & Rubinsztein, D. C. Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 7, a024240 (2017).
4. Nalavade, R., Griesche, N., Ryan, D. P., Hildebrand, S. & Krauß, S. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 4, e752 (2013).
https://doi.org/10.1038/s41556-024-01414-x责编|探索君
排版|探索君
文章来源|“BioArtMED”
End