
在金属锌负极上构建可靠的固体电解质中间相(SEI)是稳定的锌(Zn)基水系电池的必要条件。然而,同时发生的Zn2+(H2O)6溶剂化团簇的吸附(捕获)和去溶剂化给SEI的设计带来了动力学和稳定性的挑战。
成果简介
在此,复旦大学赵东元院士和晁栋梁教授等人展示了一种串联化学策略来解耦和加速在内亥姆霍兹层的Zn2+团簇的同时吸附和去溶剂过程。其中,一个具有串联亲水-OH和疏水-F基团的电化学组装的介孔孔二氧化硅作为Janus介孔加速器,以快速稳定Zn2+的还原反应。同时,结合原位电化学数字全息术、分子动力学模拟和光谱表征,发现-OH基团捕获Zn2+来自电解液体相的团簇,然后是-F基团排斥配位的H2O分子在溶剂化壳层中实现串联离子还原过程。所得到的对称电池分别在4和10 mA cm-2的高电流密度下,在8000和2000小时内实现了高度可逆性。此外,串联化学在Zn//VO2和Zn//I2电池中的可行性得到进一步证明,并可能适用于其他水系金属离子电池。
相关文章以“Tandem Chemistry with Janus Mesopores Accelerator for Efficient Aqueous Batteries”为题发表在J. Am. Chem. Soc.上。
研究背景
可充电锌基水系电池(ZABs)是“超越锂离子”储能技术之一,因其本质安全性、负担得起的低成本和环保性而受到广泛关注。然而,锌沉积/剥离的传质动力学缓慢源于去溶剂化过程的高能垒、锌腐蚀和H2O产生的析氢反应(HER),使ZABs的电化学性能变差。因此,开发一种多功能固体电解质界面(SEI),能够在负极抑制HER的同时表现出最佳动力学,但仍然面临重大挑战。其中,负极沉积反应的动力学主要受溶剂化阳离子的吸附和随后在SEI处的去溶剂化过程的控制。有效捕获溶剂化阳离子可减少界面极化和浓度极化,促进均匀离子分布和均匀沉积。同时,具有低能垒的去溶剂化过程应抑制电化学极化和溶剂降解,但溶剂化的Zn2+团簇的捕获和脱溶剂过程的启动通常在亥姆霍兹内部层内重合,这些过程本质上需要两个截然不同的SEI属性。一方面,与水分子配位的锌离子团簇容易被亲水表面截留,导致溶剂分子降解和HER。另一方面,疏水表面有助于剥离溶剂化Zn2+中的活性水团簇,但会引起离子分布不均匀和局部锌沉积。表面性能要求的这种内在矛盾对ZAB的SEI设计构成了实质性障碍。
为了解决Zn2+吸附脱溶剂化过程效率低下的问题,人们已经付出了巨大的努力来构建SEI的微观结构.这些努力包括各种材料,包括有机化合物,无机非金属材料,金属纳米粒子和多孔纳米颗粒。尽管付出了这些广泛的努力,但这些方法中的大多数都完全依赖于单一功能的SEI设计,而忽略了离子还原过程中不相容吸附和脱溶剂化的优化。此外,具有无序堆积界面的不合理SEI设计导致有机或无机化合物的高曲折和堆积,拉长了离子转移路径并降低了电荷转移电阻。因此,将离子传输的方向限制在SEI内以最大化地提高Zn2+的吸附(捕获)效率和促进脱溶(排斥)动力学至关重要。
图文导读
Janus mSiO2的合成和表征
Janus介孔SiO2的合成是通过电化学辅助的自组装过程(如图1a),包括以锌基底为工作电极的两个阶段。首先,将锌底物浸没在由四乙基正硅酸盐(TEOS)、三甲氧基(3,3,4,4,5,5,6,6,7,7,8,8,8,-十氟辛基)硅烷(PFOTMS)、硝酸钠、十六烷基三甲基溴化铵(CTAB)和盐酸组成的溶液中。采用恒流在正极生成氢氧根离子,同时控制不同硅烷的水解动力学,这触发了PFOTMS的优先自缩聚,并导致在Zn表面形成表面活性剂模板化的介孔二氧化硅纳米通道。在恒定电场作用下,由TEOS和CTAB组成的胶束在F-mSiO2中自组装进行界面和二次沉积,在−F富层上形成一个富含−OH的层。通过在Cu和ITO玻璃等不同基材上生产这种中间相,进一步证明了合成方法的普遍性,通过在特定电流下调制电沉积时间,还可以调整中间相的厚度。因此,可以成功合成具有上-OH和下-F基团的Janus介孔SiO2中间相,并将其作为稳定金属锌负极的人工界面层。
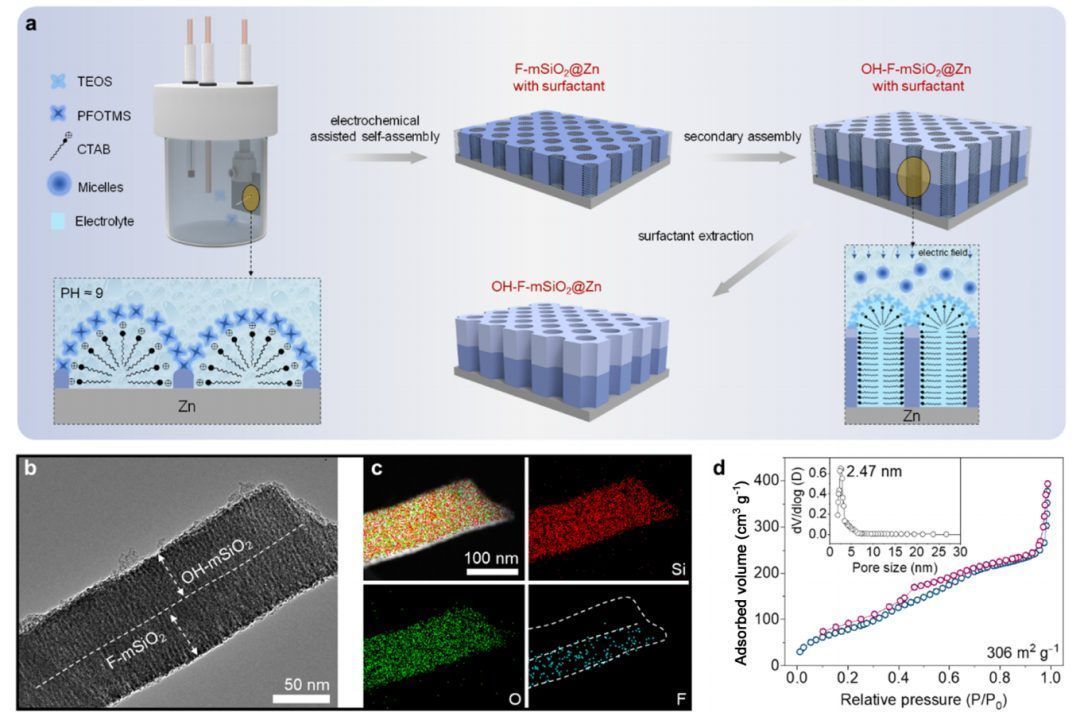
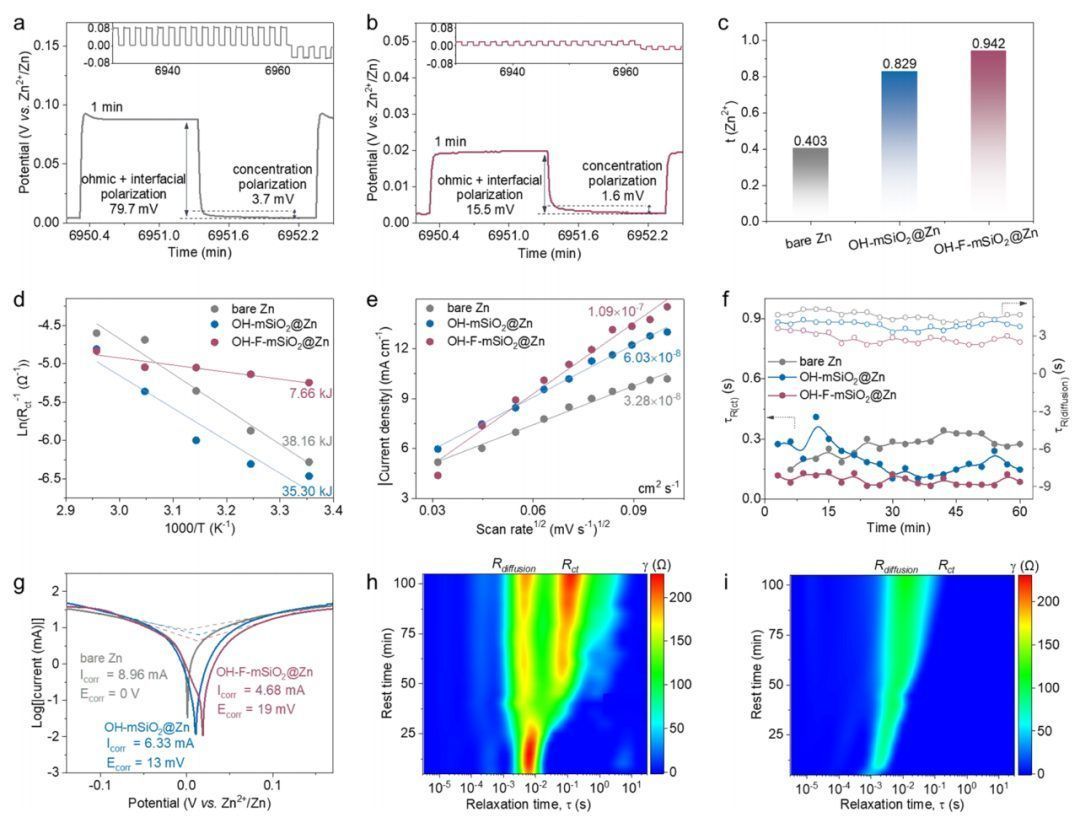
动力学和热稳定性
为了研究串联化学以及Janus介孔界面的动力学和界面化学的影响,作者制备并测试了一系列控制界面,包括裸锌负极、单亲水-OH界面(OH-mSiO2@Zn)和疏水性-F界面 (F-mSiO2@Zn)。值得注意的是,Zn2+的快速界面动力学,包括离子电荷转移和沉积动力学,对于高倍率ZAB至关重要。与接触角为85.9°的裸锌电极相比,OH-mSiO2@Zn(59.9°) 和OH-F-mSiO2@Zn(68.7°)电极由于其极性亲水基团而显示出较小的与电解液的接触角。亲水界面有助于降低电荷转移阻抗和增加离子电导率。此外,介孔通道有效捕获Zn2+,将浓差极化从3.7降低到1.6 mV,欧姆和电化学极化从裸Zn的79.9 mV降低到OH-F-mSiO2@Zn的15.5 mV。值得注意的是,具有阴离子排斥的介孔相促进了Zn2+选择性从裸锌的0.403提高到OH-mSiO2@ Zn的0.829和OH-F-mSiO2@Zn的0.942。综上所述,OH-F-mSiO2串联化学的界面调控不仅可以作为介孔加速器来促进吸附,还提高作为Zn2+的脱溶剂动力学。
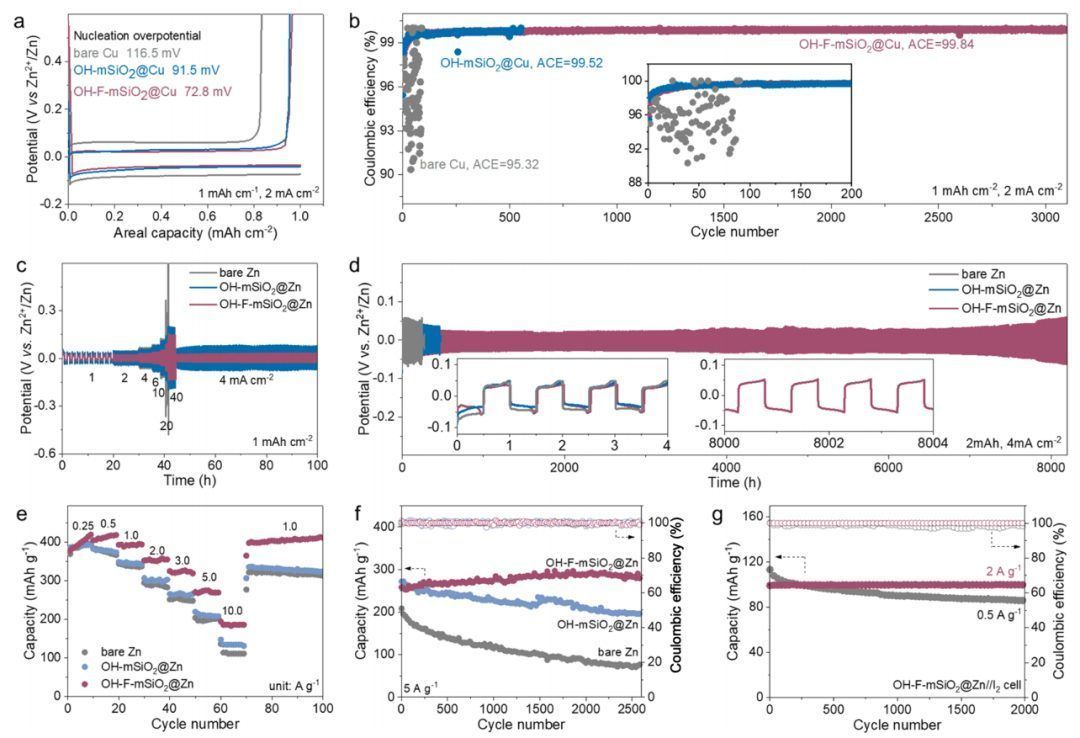
电化学性能验证
图3a中不对称Cu//Zn电池与裸Cu(116.5 mV)和OH-mSiO2@Cu(91.5 mV)相比,OH-F-mSiO2@Cu(72.8 mV)的成核过电位更低,且裸Cu//Zn电池的平均库仑效率(ACE)较低,仅为95.32%,由于短路而在不到100个循环内失效(图3b)。相比之下,ACE可以通过介孔界面来提高:OH-F-mSiO2@Cu//Zn电池在3000个循环中达到了99.83%的ACE,这意味着副产物的形成有限。均匀的锌沉积可以归因于Zn2+在绝缘的Janus介孔界面中有利的三维扩散。相比之下,裸锌的二维扩散容易导致锌枝晶的生成。图3c中的OH-F-mSiO2@Zn对称电池显示了不同电流密度的最低极化电压,表明其快速的锌沉积/剥离动力学。此外,在4和20 mA cm−2的高电流密度下,电池的长循环寿命分别超过8000和800小时(图3d)。
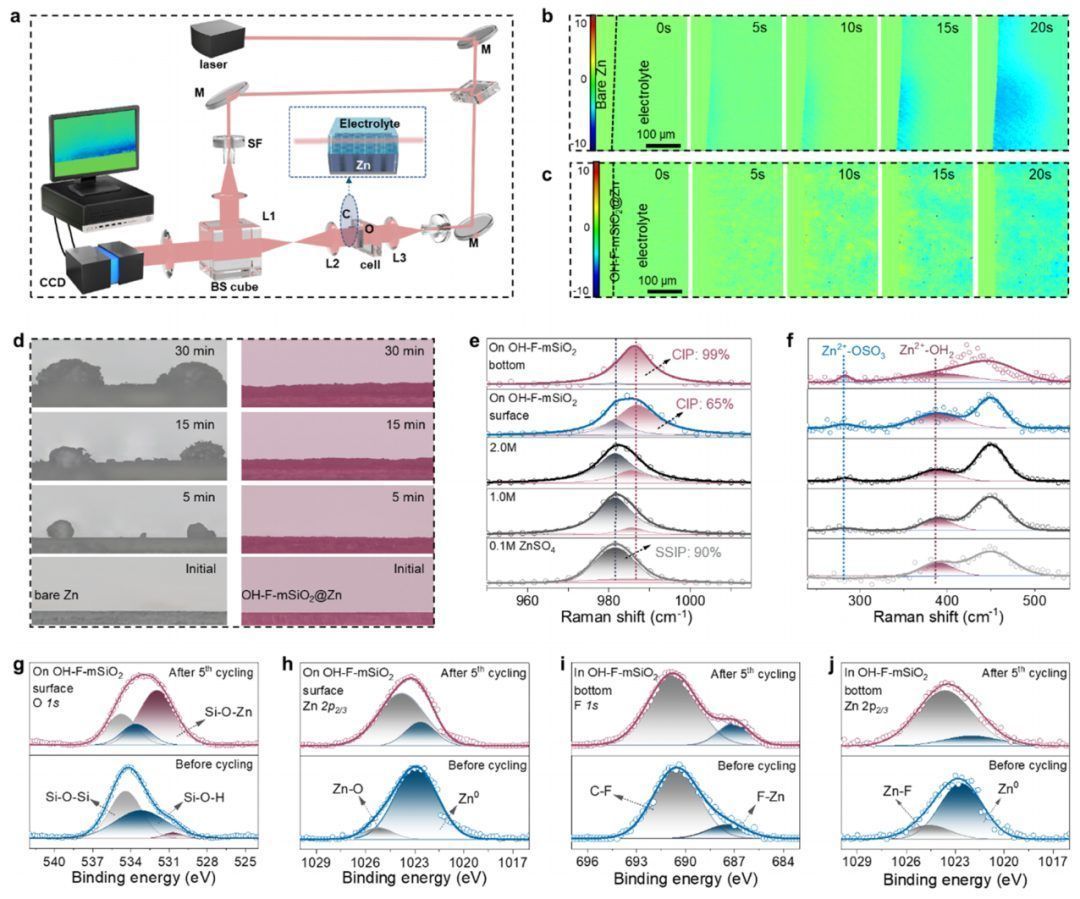
溶剂化结构表征
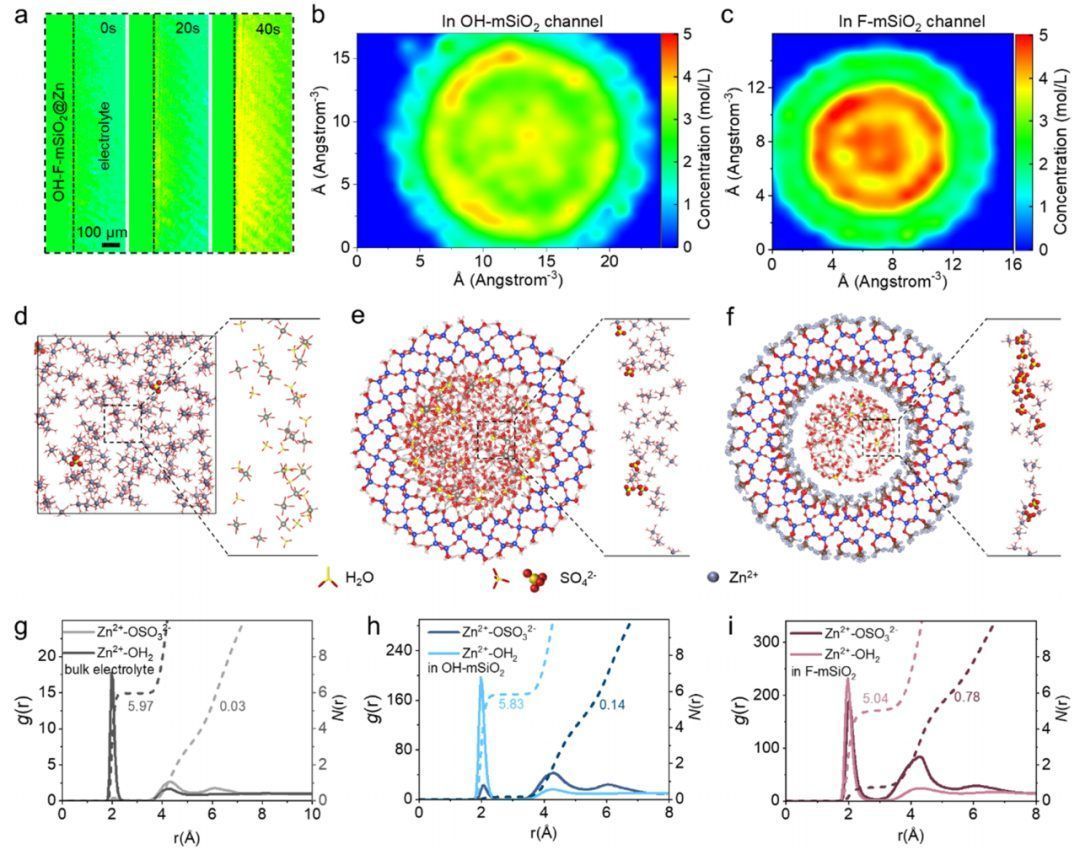
为了阐明Janus介孔加速器串联化学的潜在机制,作者对界面进的物理化学特征进行了深入的研究。利用原位电化学数字全息技术(EDH)(图4a),通过测量光路长度分布,监测了电极/电解液界面初始锌离子还原过程中的离子浓度的演化。在电化学还原20 s后,记录了明显的局部颜色变化,表明Zn2+有明显的浓度梯度(图4b)。相比之下,分布在上中孔通道中的Zn2+捕获基团可以主动、均匀地从电解液体相中吸收Zn2+(图4c)。这一有利条件是实现无枝晶锌沉积的关键前提,且原位光学显微镜结果验证了上述在OH-F-mSiO2@Zn表面的无枝晶锌沉积(图4d)。
介孔加速器模拟
将OH-F-mSiO2@Zn负极浸入电解液中后,原位EDH记录了明显的且均匀的颜色变化(图5a),这归因于串联化学的界面通过静电力从电解液体相中捕获Zn2+,因此Zn2+的界面浓度逐渐增加。因此,分子动力学(MD)模拟被应用于定量研究Zn2+在介孔通道内的再分配,并提供了原子水平的见解。模拟结果表明,极性-OH基团有效地捕获了Zn2+,而Zn2+在OH-F-mSiO2表面附近积累。如图5b所示,Zn2+的浓度在富-OH区域模拟超过4 mol/L,是电解液体相中的2倍,从而实现了一个局部的高浓度微环境,这证实了界面锌离子再分布的观察。相比之下,疏水富-F区将排斥溶剂化Zn2+,得到了更高浓度的Zn2+(5 M)和去溶剂的Zn2+结构。
总结展望
综上所述,作者通过在设计复杂的Janus介孔加速器中诱导它们的异步发生,提出了一种串联化学策略,来解耦和加速Zn(H2O)62+团簇关键的捕获和脱溶过程。其中,介孔二氧化硅可以与上亲水性-OH和底疏水性-F基团进行电化学组装。Zn(H2O)62+团簇可以在富-OH被捕获,然后在富-F区进行异步脱溶过程(方案1b)。原位数字全息和电化学过程分析显示,Janus介孔相的界面极化降低和脱溶动力学加快(活化能为7.66 kJ mol−1,为裸锌的20%)。分子动力学模拟和光谱分析提供了进一步的原子水平的见解,有趣的是精心设计的锌负极表现出特殊的稳定性和动力学,即在电流密度为4 mA cm−2的情况下,也能实现超过8000 h的长循环,且与Janus介孔孔加速器的串联化学可以激发其他金属基电池的成功。
文献信息
Lipeng Wang, Bao Zhang, Wanhai Zhou, Zaiwang Zhao, Xin Liu, Ruizheng Zhao, Zhihao Sun, Hongpeng Li, Xia Wang, Tengsheng Zhang, Hongrun Jin, Wei Li, Ahmed Elzatahry, Yasser Hassan, Hong Jin Fan, Dongyuan Zhao,* and Dongliang Chao*, Tandem Chemistry with Janus Mesopores Accelerator for Efficient Aqueous Batteries, J. Am. Chem. Soc.. (2024).