引言
实际的科研活动中,组学产品的快速更新换代导致研究员中对相关技术产品并不是太了解,因此除了要了解研究趋势外,还要精准匹配研究需求所对应的组学产品;引用赵立平教授的观点“创新是好的研究和文章的前体”:创新除了研究问题的深入洞悉和独特角度外,还包括各种技术不断迭代的组学产品。“好的实验设计是研究的地基,成熟的组学手段是研究的脚手架”,匹配研究的组学产品可以事半功倍的解析科研问题,为一篇好文章提供稳定可靠的数据。同时,现在各种AI工具和基础模型可高效助力研究者的数据分析,也可为文章的撰写提供各种便利,大大缩短从“idea”到“article”的时间间隔。同时,现在各种AI工具和基础模型可高效助力研究者的数据分析,也可为文章的撰写提供各种便利,大大缩短从“idea”到“article”的时间间隔。但是就像施一公说的“AI可以帮科学家搬砖,但AI并不会盖房子”,AI可以协作,但一篇文章从思路到成稿的核心还是科学家本身。
文章设计“流水线”
ORIGINGENE
1、好的实验设计
变量设置、分组设置;好的实验设计是一篇好文章的基础;一定要提前定好研究主题,变量设置和分组设置以及各种表型信息的搜集,以及根据生理生化结果及时调整实验设计;同时创新性的实验设计也可减小被抢发的概率;好文章“和而不同”:可以参考特定期刊的高分文章,也可以模仿其研究思路和实验设计,但一定要根据自身研究的特征和具体数据进行创新性的改造,即使在结果不符合预期的基础上也要能找到分析数据和实验数据的核心要点。
2.精准的数据分析
数据可以多但不能杂:要明确每个分析数据的作用,以及数据之间的关联;数据分析“稳中求快”,在保证数据没有问题的基础上,一定要尽快根据实验设计(这时候就体现出一个好的实验设计是多么重要了)快速的筛选海量数据中有价值的或者说跟研究高度相关的数据(基因、通路、功能、基因簇等)。这里可以稍微参考上篇技术贴:技术介绍 | 如何从转录组数据中筛选候选基因 (qq.com)
3.完备的实验验证
新发现的验证:明确后续的分析流程和验证流程,将新发现做实。
4.“他山之石,可以攻玉”
前人研究和自身数据是“相辅相成”的:不能一味参考前人研究否能自身的实验和分析数据,同时也不能过于看重自身分析数据而忽略前人的研究成果;在自身分析数据和实验数据的基础上,通过“文本匹配”,筛选相似的研究都有哪些成果,对这些成果进行有效汇总,然后跟自身结果进行整合,整合后的结果就可以更高效地进行后续的实验或数据分析安排。
5.文章格式
现在AI已经成熟的基础上,上述实验设计、数据分析和实验验证走通的基础上,一篇格式严谨的文章产出会相对快很多,可以借助AI进行特定期刊文章格式的校正和修改,甚至某些特定篇幅的润色也可用AI进行解决。
实验设计
ORIGINGENE
进行实验设计前首先要明晰研究领域的发展方向和发展趋势,然后结合自己的研究条件和前期成果,专注于细分的研究方向;创新性的研究:即所做的研究和相关发现,一定是前人没有做过的,或者是前人研究中有所遗漏的;常见的思维是探索新的物种,组织,细分方向甚至要开拓这个科学问题的边界(上下游研究、ncRNA调控等),同时充分研究的基础上,可以对这个科学问题提出自己不同的见解(例如上述问题是否不是常规基因调控,而是逆转录病毒转座子调控呢?)。
综上,一个好的实验设计的第一要义是“创新性”,可以参考,可以学习,但一定要“和而不同”“推陈出新”,在前人数据基础上,进行创新性的研判。可以参考最新的综述和相关研究:例如本次研究的重点是RNA修饰,就可以参考如下综述进行研究思路的梳理和扩展:高分综述 | RNA修饰对免疫代谢的调节会进一步影响免疫反应 (qq.com);
基础设计
ORIGINGENE
在创新性的基础上,一定要提前构建好整篇文章的研究路径:主要解决如下问题:
1.要怎样设置分组?
实验组、对照组、处理组(处理类别、浓度、条件等)、生物学重复(转录组建议5个以上,有些组学会有不同的情况)。例如针对不同的癌症研究场景,就可以设置出如下不用的分组:癌症和正常;癌症转移与否;耐药与否;癌症发展阶段;癌症亚型;治疗响应与否;肿瘤微环境;跨癌症比较。
2.要做哪些组学?
要根据研究需要进行哪些组学检测和测序,并不是越多越好,转录组、蛋白组、代谢组、表观组、单细胞组、空间组还有微生物组,可以根据研究方向的设定,定好大方向上的组学技术。
3.样品采集?
在保证稳定样品采集后,可以适当多采集一些样品,用于后续实验验证或者出现新的研究结果需要增加新的组学检测,以备不时之需。
数据分析
ORIGINGENE
针对数据分析,通常包括“已知”和“未知”。已知数据库的分析逻辑是:拿到分析的序列信息,和一个数据库做比较,分析序列和数据库中已知的东西相似性超过一定阈值,就定义为数据库中的东西,不能和数据库中足够像的序列就被“遗弃”掉,全部都做了已知定义后再对它进行分析;这样有可能会把有价值的未知数据丢弃掉,因此就需要采用一些创新型的分析软件和流程,对这些“未知数据”进行系统性的分析,以期拿到有价值有创新点的研究数据。“未知”分析中,会采用一些前沿或者个性化的软件和算法研究去拓展组学数据的边界,发现更多有价值的“未知”数据。上述分析更多的是单“因子”分析,即从“点到点”的思维分析基因和基因集,但对于生物来说,生命活动是一个复杂的系统,需要进行系统性的研究“因子”之间的复杂网络关系。
1.数据“已知”分析
进行组学研究的时候,通常会参考一些数据库(GO/KEGG等)进行序列注释和分析,这些数据库很完整,有系统性的功能架构,也有现成的分析软件和脚本,通常来说可以拿到很多“已知”的数据结果;对“已知”数据根据因素和变量研究,通常为筛选基因或筛选基因集的分析(ORA),通过设定阈值参数筛选参数好的差异基因和基因集(差异基因和显著富集的pathway等),具体流程可以参考上篇文稿:技术介绍 | 如何从转录组数据中筛选候选基因 (qq.com)。
2.数据“未知”分析
数据“未知”分析,需要借助新的软件和算法流程去挖掘数据深层次蕴含的信息,同时在“已知”分析结果的基础上,进行合理的推演和预测:如下研究中就采用了新的研究软件和研究思路,发现了很多以前没有发现的线粒体circRNA。(Liao,2022) ,具体文献解读:署名文章 | 线粒体编码的环状RNA在植物中广泛存在并可翻译 (qq.com)。

图 使用MeCi鉴定玉米和拟南芥中的mcircrna
3.数据“系统”分析
针对“已知”数据进行因素和变量研究的同时也要进行系统性的研究。系统性的研究对于找到真正至关重要的基因和研究方向至关重要,筛选到候选基因后,需要考虑到这些基因在生物复杂系统中的位置和作用,还需要考虑这些候选基因上下游调控及本身的结构改变(m6A修饰、RNA转运、转录因子调控等;因此需要从复杂系统(真实的生物系统过于复杂,需要进行降维,研究其中的几个关键维度)中的关键维度信息,借助已知数据库进行各种维度信息的注释和关联。(生物复杂系统介绍参考:Cell | 逆转录转座子介导的脊椎动物髓鞘形成机制 (qq.com))。

图 RNLTR12-int调控SOX10与MBP启动子的结合
基础模型和微调模型
ORIGINGENE
常规的计算生物学机器和深度学习主要是专注于非常具体的任务,这些模型可以对DNA、RNA和蛋白质的结构和功能进行复杂预测,并能够根据进化原理生成新的生物分子。
在上述框架和模块基础上,延伸出的概念“基础模型”:其主要场景为借助预训练数据构建基础模型,然后在基础模型上根据研究方向进行模型微调,用于预测“未知”数据并进行初步定义。更复杂的AI算法,可以“解释”生物数据的模型,进而帮助指导生物分子设计;类似于GPT-4的AI方法现在正在产生“基础模型”(Eisenstein,2024);基础模型的特征是在大量未注释的数据集上进行“预训练”,然后可以解决新的问题;
1.机器学习和基础模型
深度学习可以用大数据集挖掘数据的隐藏结构,并进行预期预测;机器学习的工作流程可以分为四个步骤:数据预处理、特征提取、模型学习和模型评估(Angermueller,2016)。在被用于预测之前,模型需要进行训练,包括自动调整模型的参数以提高其性能(Greener,2022)。

图 选择模型的决策树_选择训练机器学习方法
如下展示为一些常见的生物基础模型:
ESM3:EvolutionaryScale 发布,可以利用大量的结构、序列和功能数据来指导蛋白质设计,进而指导不同的分析和设计任务;ESM3采用有注释的数据(28亿个蛋白质的多种类型数据)串联在一起,以推断和概括现实世界的进化过程(Hayes et al.)。
BigRNA :Deep Genomics开发:整合了多种机器学习的预测能力,同时又具有对交叉生物过程预测的能力,能做出更准确的预测,可以预测出从未见过的生物分子;BigRNA模型可以准确预测以前从未遇到过的基因结构和表达水平特征(Celaj et al.),
Atomic AI:Stephan Eismann开发,模型可以预测基于RNA的药物及其靶点的结构和物理化学性质;模型通过引入广泛的化学映射实验,基于化学图谱数据,Atomic AI可以准确预测RNA的二级和三级结构。
scFoundation:BioMap发布,基于5000万个人类细胞的单细胞数据,基于Transformer架构通过捕捉不同细胞类型和状态下基因之间的复杂关系,进行RDA建模;模型可以用于细胞类型的聚类,并预测细胞对不同药物的反应。
GPT4:作为一种基础模型,是通过大量互联网文本进行预训练(NLP建模);生物学的基础模型遵循相同的逻辑,也是建立在LLM框架基础上,区别是从“人类语言”变成了“DNA、RNA、蛋白质”(序列结构建模)。生物基础模型的构建过程与GPT类型,需要成规模以及多样化的数据,使用来自众多物种或细胞类型的数据可以最大限度地减少潜在偏差;基于LLM的AI模型使用“参数”来分类和处理数据,参数的数量决定了结果分析的质量,例如GPT4使用了超过1万亿个参数,同样的,生物基础模型为了实验其预测的精准度,同样需要实现这个规模的参数。
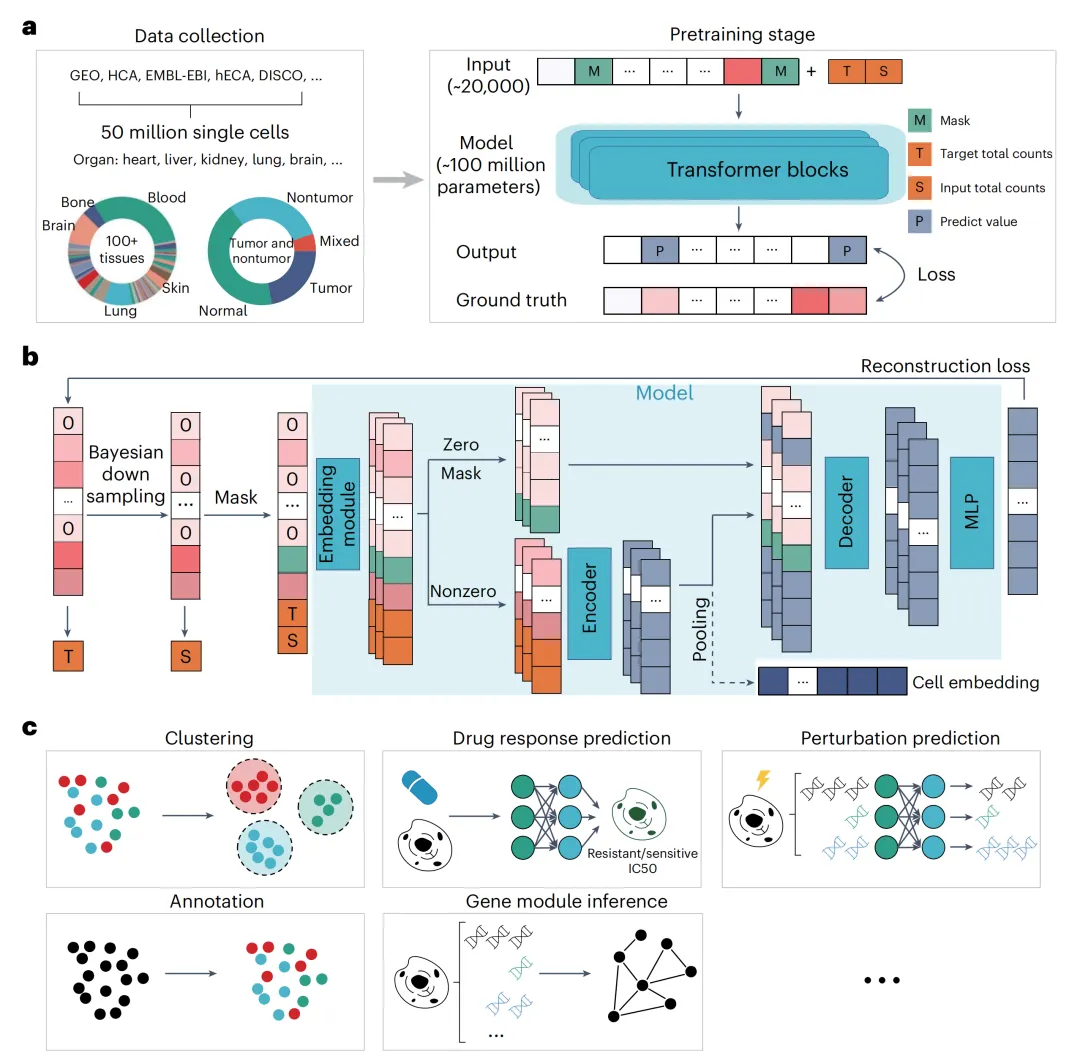
图 scFoundation训练框架的示意图概述
2.微调模型
生物基础模型是一种基础设施,一般情况下不需要直接训练基础模型,一般是在开箱即用的基础模型上,进一步专门训练,使用更小的、更具针对性的数据集来进行定向训练,生成微调模型(Fine-Tuned Model),如微调后的模型可以用于解决某个具体的生物学问题,例如设计新的蛋白质结构、预测基因编辑效果或发现新的治疗靶点等。xTrimoPGLM是一个蛋白质模型,其具有1000亿个参数,可以预测蛋白质的结构、稳定性和与其他分子靶标相互作用,进而可以设计具有特定功能和特性的定制分子。Sanofi通过定制化的数据集对xTrimoPGLM基础模型进行微调,进而优化蛋白质结构预测、抗体-抗原相互作用预测,甚至RNA疗法设计等领域中的具体任务。

图 xTrimoPGLM选择ColAbFoldDB和Uniref90两个数据集预测后的蛋白质分类结果
如下简单介绍下生物基础模型的微调流程:针对肝癌的转录调控特征以及药物靶点的预测作为研究出发点。
(1)选择基础模型:根据自身研究需求选定基础模型,基础需求抽象关键词为“生物学过程”和“药物靶点”,因此可以使用BioBERT 和 MedBERT(基础模型是在大规模生物医学文献和数据库上进行的训练,能够处理基因名称、疾病、药物信息等生物医学知识)进行文献挖掘;基础模型选择的基本逻辑是:数据量(尽可能选择大参数的模型)、相似度(选择基础模型的预训练数据要和研究方向的数据非常接近)。
(2)数据准备和预处理:选择适合模型的数据集,可以使用转录调控数据集(RNA、表观修饰、单细胞等;TCGA、GEO等数据库)提取转录调控数据,药物-靶点数据库(DrugBank等)提取已知的药物靶点和药物分子。预处理:数据去噪(尤其是跨数据集整合时)、数据标注(跨数据类型时,需要对不同组学的数据进行标注,从而将不同组学数据关联起来),保证数据质量。
(3)模型加载和微调:选定深度学习框架中加载这些预训练模型;为了优化模型在本研究方向的预测效率和准确率,需要进行微调,微调逻辑主要是两个:冻结部分模型参数和加入研究方向特异参数
a)冻结参数:保留基础模型中的一些通用特征提取层,冻结权重,只微调输出层或特定任务相关的网络层。
b)特异参数:将肝癌数据集输入到模型中进行训练,微调模型权重,对于BioBERT,可以将与肝癌相关的基因、转录因子和疾病描述数据作为特异参数。
c) 小学习率:一般而言,基础模型为了保证能够适应大规模的数据并进行有效捕获输出,通常采用较大的学习率;,基础模型已经通过预训练获得大量通用参数,因此微调阶段只需要设置较小的学习率,以避免过度拟合并进行更精细化的预
(4)模型评估与优化:在微调过程中进行交叉验证,以确保模型在新数据上的表现是可靠,然后通过微调模型,可以分析肝癌组学样本中的转录调控数据特征,例如识别关键转录因子,构建肝癌特异性的转录调控网络。

图 模型加载和微调简单示例
实验验证
ORIGINGENE
1.构建“系统生物学模型”
实验验证的目的就是把数据分析结果中的“因果关系”做实,研究者在设计实验时,不能局限在描述性的层面,而是要构建一个系统性的结果(上下游串联,且有实验数据支撑)。例如某个转录因子调控某个基因的表达,那就要证明如下信息:转录因子可以和这个基因的启动子区域结合(序列和空间上),一般用到ChIP验证序列结合,用荧光素酶基因表达验证空间上结合;转录因子确实影响了基因的表达,对转录因子基因进行敲除过表达,或者引入药物拮抗转录因子引入外源合成转录因子,定量查看基因转录本的表达;通过上述路径还原了“转录因子-调控基因-转录本水平”的调控路径。在上述基础上可以进一步拓展研究的边界:例如转录因子又会受到哪些基因的调控?基因的表观修饰水平是否会影响转录因子的集合?基因转录本对下游的调控是怎样的?除了本研究的表型外,上述基因转录本还会调控哪些表型?拓展边界后需要根据数据和前期验证结果研判具体该做哪些验证实验或增加组学分析,抓住一条主线,构建上下游调控机制。上述流程就跟黏菌在迷宫中寻找燕麦一样,需要搭建最优的实验验证路线。

图 黏菌搭建起了多个“燕麦”之间的最优路线
2.转录组标准实验验证流程
验证RNA的调控机制,需要分别验证RNA表达的真实性,并对RNA进行体内和体外功能验证,其核心流程是:验证RNA真实存在-验证RNA的表达水平-验证RNA的功能。
具体的实验类型可以参考如下文章:lncRNA专题 | lncRNA的功能验证流程 (qq.com)
如下案例为一篇文章中的实验验证流程:组学数据分析结果显示LncRNA对应的多肽研究中,lncRNA-00998编码的SMIM30促进SRC/YES1膜锚定并激活MAPK通路;lncRNA-MCM3AP-AS1吸附miR-194-5p,使得FOXA1表达升高,促进肿瘤发展(Cha,2024)。实验验证的目的为通过实验设计验证上述分析和推理出的转录调控轴。

图 研究转录后调控因子UPF1对特定lncRNA的影响
3.系统生物学验证流程
上述流程是从线性交付验证上下游的基因和转录本调控,但实际的生物系统中,上下游的“因子”是相互调控的,是一个网络化的调控系统,因此,高分文章中对组学分析的结果进行验证的时候,多是以“网络化”的验证流程,串联起多个“因子”的相互调控关系的(Xu Y,2023)。如下流程中需要证实:Hippo通路中核心基因LATS1的m6A甲基化对乳腺癌细胞增殖和代谢的调控机制、METTL3通过诱导LATS1 mRNA的m6A甲基化,并可以被YTHDF2识别来发挥致癌活性、YAP/TAZ的磷酸化水平和定位-Hippo通路的活性受到乳腺癌细胞中LATS6的m6A调节、m6A修饰调节通过Hippo途径因子LATS6在乳腺癌中的增殖和糖酵解代谢过程、METTL3-LATS1-YTHDF2通路通过激活Hippo通路中的YAP/TAZ在乳腺癌的进展中起重要作用。需要通过如下实验和补充分析流程逐步验证上述调控路径:荧光定量PCR;细胞增殖、凋亡、迁移和侵袭测定;免疫共沉淀测定;MeRIP-seq;代谢组;转录组;SRAMP(预测m6A甲基化位点);RNA pull down;RIP-qPCR。

文章撰写
ORIGINGENE
文章撰写,作为一个非英语母语者的撰写者,除了要考虑文章图表、文章结构和详略得当的内容外,还要注意语言问题(词不达意、语法描述等),如下主要描述在文章撰写中AI+的常见操作;AI分类中Research Article write AI属于Genera/Personal Assistant(通用/个人助手类)AI下的分支,如下会简单介绍一些常用的工具。AI工具可以用于规划文章结构和帮助改写片段甚至进行文章润色;
1.Hyperwrite
Hyperwrite(基于ChatGPT进行的微调模型)针对不同场景提供了不同的写作工具, Hyper可以根据设定好的论文主题,初步生成结构化的大纲,便于进行想法组织,为论文初步生成清晰路线图(引言,Highlight和结论);Hyper针对Scholar场景下提供了可以根据检索到的数百万篇学术文章来进行实时总结,提供准确且有最新论文支持的结果,分析这些文章并收集关键点、论点和发现,并使用这些信息来完成研究请求和写作任务;

图 Hyperwrite操作界面
2.HIX.AI
HIX.AI(也是一个GPT微调模型,只要是文本生成基本绕不过去GPT)是一个AI写作助手,除了可以生成文本外,还可以进行错误校正、总结内容,查重以及进行英语语法的润色。其相对于基础模型的微调点主要在于“Prompt engineering”和生成后内容的“Academic Editing”

图 HIX AI的操作面板
3.Web of Science研究助手
Web of Science研究助手(基于生成式AI)经过改进的检索功能可以快速引导研究人员找到相关内容;还可以处理复杂任务:了解每个主题、寻找适合投稿的期刊以及完成一篇文献综述;研究助手还能利用丰富的被引文献数据揭示研究人员引用论文的方式以及原因。

图 web of science研究助手提供智能发现和任务引导功能
利用基础AI来突破研究和学习的界限,采用商业预训练的LLM(大语言模型)作为信息检索和增强框架的一部分,可以快速定位某一领域的顶尖研究者,并链接到详细个人资料;利用动态可视化工具发现文章间有意义的关联。

图 主题图谱揭示了相关课题领域

图 语境化提示进一步探索新领域
4.Zotero+obsidian
但是现阶段AI依旧存在一些短板,例如上下文语义错位严重,无法完成专业概念阐述问题以及跨学科的思维发散,AI也是一种会犯错的工具,不可过分依赖;也许尝试了各种AI工具后,发现Zotero+word+obsidain才是最简洁也最好用的工具组合。

图 Zotero文献管理界面
参考文献
Eisenstein, Michael. “Foundation Models Build on ChatGPT Tech to Learn the Fundamental Language of Biology.” Nature Biotechnology, vol. 42, no. 9, Sept. 2024, pp. 1323–25. www.nature.com, https://doi.org/10.1038/s41587-024-02400-2.
Hayes, Thomas, et al. Simulating 500 Million Years of Evolution with a Language Model. bioRxiv, 2 July 2024, p. 2024.07.01.600583. bioRxiv, https://doi.org/10.1101/2024.07.01.600583.
Celaj, Albi, et al. An RNA Foundation Model Enables Discovery of Disease Mechanisms and Candidate Therapeutics. bioRxiv, 26 Sept. 2023, p. 2023.09.20.558508. bioRxiv, https://doi.org/10.1101/2023.09.20.558508.
Liao, Xun, et al. “Mitochondrion-Encoded Circular RNAs Are Widespread and Translatable in Plants.” Plant Physiology, vol. 189, no. 3, June 2022, pp. 1482–500. 7.4, https://doi.org/10.1093/plphys/kiac143.
Greener, Joe G., et al. “A Guide to Machine Learning for Biologists.” Nature Reviews Molecular Cell Biology, vol. 23, no. 1, Jan. 2022, pp. 40–55. www.nature.com, https://doi.org/10.1038/s41580-021-00407-0.
Angermueller, Christof, et al. “Deep Learning for Computational Biology.” Molecular Systems Biology, July 2016. world, www.embopress.org, https://doi.org/10.15252/msb.20156651.
Applications of Machine Learning and Deep Learning on Biological Data | Faheem Masoodi, Mohammad Quasim, Syed Nisar Hussain Bukhari, Sarvottam Dixit, Shadab Alam | Download on Z-Library. https://zh.z-lib.gs/book/24575071/fd8dd8/applications-of-machine-learning-and-deep-learning-on-biological-data.html. Accessed 27 Sept. 2024.
ATOM-1: A Foundation Model for RNA Structure and Function Built on Chemical Mapping Data | bioRxiv. https://www.biorxiv.org/content/10.1101/2023.12.13.571579v1. Accessed 22 Sept. 2024.
Vert, Jean-Philippe. “How Will Generative AI Disrupt Data Science in Drug Discovery?” Nature Biotechnology, vol. 41, no. 6, June 2023, pp. 750–51. www.nature.com, https://doi.org/10.1038/s41587-023-01789-6.
Ille, Alexander M., et al. “Generative Artificial Intelligence Performs Rudimentary Structural Biology Modeling.” Scientific Reports, vol. 14, no. 1, Aug. 2024, p. 19372. www.nature.com, https://doi.org/10.1038/s41598-024-69021-2.
Ghosh, Tanay, et al. “A Retroviral Link to Vertebrate Myelination through Retrotransposon-RNA-Mediated Control of Myelin Gene Expression.” Cell, vol. 187, no. 4, Feb. 2024, pp. 814-830.e23. 64.5, https://doi.org/10.1016/j.cell.2024.01.011.
Cha, Hyunho et al. “Role of UPF1 in lncRNA-HEIH regulation for hepatocellular carcinoma therapy.” Experimental & molecular medicine, 10.1038/s12276-024-01158-6. 1 Feb. 2024, doi:10.1038/s12276-024-01158-6.
Xu, Youqin et al. “The N6-methyladenosine METTL3 regulates tumorigenesis and glycolysis by mediating m6A methylation of the tumor suppressor LATS1 in breast cancer.” Journal of experimental & clinical cancer research : CR vol. 42,1 10. 7 Jan. 2023, doi:10.1186/s13046-022-02581-1