二甲双胍是经典的降糖药物,临床应用已有60多年,且具有降糖之外的多种益处,更多治疗潜力还在不断展现中,素有“神药”之誉。这种源自天然草药山羊豆的药物,为何能拥有如此宽泛的功效?尽管目前为止二甲双胍的作用机制尚未完全阐明,但众多证据支持,激活腺苷酸活化蛋白激酶(AMPK)信号通路是二甲双胍发挥降糖以及降糖外多效性作用的核心机制。那么,AMPK究竟是什么?具体又怎么激活?为帮助临床医生了解相关机制,本文特邀哈尔滨医科大学附属第一医院医院匡洪宇教授,为您解读开启二甲双胍神奇之处的这把“密钥”。

一、了解AMPK:人体内能量代谢“总开关”,神奇功能“一箩筐”
AMPK,全名5′-AMP-activated protein kinase,顾名思义,这是一种能被单磷酸腺苷(AMP)激活的蛋白激酶。作为一种可感受能量代谢变化的“古老感受器”,AMPK广泛存在于从酵母到哺乳动物的真核细胞生物中,承载着极其重要的代谢调节作用[1,2]。
01
AMPK结构及激活机制
AMPK是一种丝氨酸/苏氨酸蛋白激酶,由催化亚基α和调节亚基β、γ组成[2,3]。作为真核细胞中高度保守、最重要的能量感受器,AMPK被喻为人体内能量代谢的“总开关”,可以感知细胞内葡萄糖水平变化,当能量不足时被激活。AMPK的经典激活途径为:当细胞发生代谢性应激时,三磷酸腺苷(ATP)水平降低,AMP/ATP、二磷酸腺苷(ADP)/ATP比值增加,在上游激酶肝激酶B1(LKB1)和钙/钙调蛋白依赖性激酶激酶2(CaMKK2)的作用下,AMPK被激活(图1)[2,4]。

图1. AMPK结构和激活
以下代谢应激情况都会导致AMP/ATP、ADP/ATP比值的增加:①当细胞处于葡萄糖、氧气缺乏状态时;②药物处理后细胞呼吸链抑制时和ATP合成抑制时;③ATP消耗增加,或运动蛋白激活(如肌肉运动)时[1]。简言之,AMPK的典型激活依赖于能量的波动变化,好比手机中的“省电模式”,在能量不足时自动开启,帮助高效利用剩余能量。
02
AMPK激活开启系列“神奇”功能
AMPK激活的作用是重新调节代谢,减少合成代谢过程(减少ATP消耗),同时增加分解代谢(增加ATP产生),以恢复更有利的能量平衡。一旦被激活,AMPK可调节多种代谢过程,包括蛋白质代谢、脂质代谢、葡萄糖代谢等。AMPK激活刺激肝脏脂肪酸氧化和糖酵解,刺激骨骼肌脂肪酸氧化和葡萄糖摄取,抑制胆固醇合成、脂肪生成和甘油三酯合成,抑制脂肪细胞脂肪生成和促进脂肪分解,以及调节胰腺β细胞的胰岛素分泌等。除调节代谢外,AMPK还调节细胞功能,如自噬、线粒体和溶酶体稳态、DNA修复和免疫等(图2)[2]。

图2. AMPK激活调节多种代谢过程
AMPK的作用关乎生命与健康的多个方面,与代谢性疾病、炎症、肿瘤、衰老等密切相关。人们发现,二甲双胍、阿司匹林、小檗碱等多种“神药”发挥作用均与AMPK通路相关[1,5]。这激发了科学家们对进一步阐明AMPK在不同组织细胞及整体水平上的调节机制的浓厚兴趣。随着研究不断进展突破,二甲双胍与AMPK之间的神秘联系也逐渐清晰,下面就让我们来一起看看。
二、不走寻常路:二甲双胍激活AMPK的关键分子靶点揭秘
01AMPK非经典激活途径——溶酶体途径
21世纪初的一个里程碑研究发现,二甲双胍可通过激活AMPK来抑制肝脏糖异生[6]。然而,二甲双胍具体如何激活AMPK的分子机制一直众说纷纭。基于体外实验的既往观点普遍认为,二甲双胍通过抑制肝细胞中线粒体电子传递链的复合物Ⅰ,导致ATP降低和AMP/ATP、ADP/ATP比值增加,进而激活AMPK,即经典激活途径[7,8]。然而,后来这一猜想被质疑,因有研究发现在临床药理剂量下,二甲双胍的血药浓度远不足以增加AMP/ATP和ADP/ATP比值而激活AMPK[9,10]。那么,临床剂量的二甲双胍是否通过别的途径来激活AMPK?2022年,发表于Nature杂志的一项中国研究揭开了这个谜底[11]。
中国研究者巧妙地通过特异性分子探针方法,“钓鱼”破解了二甲双胍直接作用靶点之谜,结果发现,早老素增强子2(PEN2)是二甲双胍发挥神奇作用的直接分子靶点,是激活AMPK的中介蛋白。简言之,临床相关浓度的二甲双胍可结合γ分泌酶复合物中的PEN2蛋白,再结合ATP6AP1蛋白募集到溶酶体空泡型ATP酶复合物上,导致空泡型ATP酶变构,便于LKB1募集到溶酶体,接触并激活AMPK(图3)[11]。
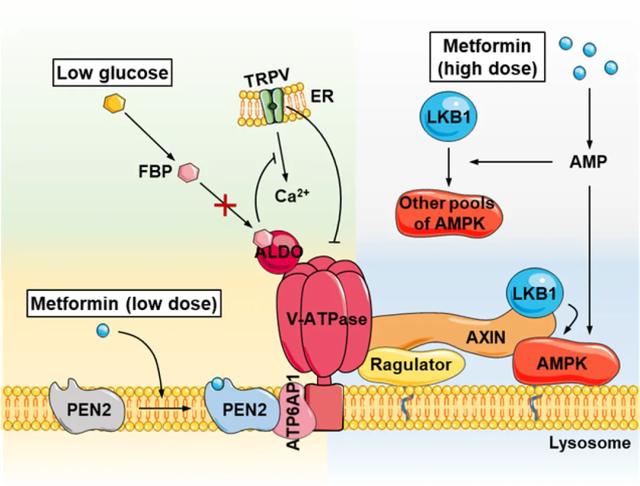
图3. 基于二甲双胍-PEN2-ATP6AP1及其他AMPK激活信号通路
这一里程碑式发现被称为“林通路”,即二甲双胍激活AMPK的溶酶体途径;该通路不依赖于AMP,但与葡萄糖饥饿、热量限制“殊途同归”。研究者认为,这条通路不需要抑制ATP生成,不会使细胞内AMP、ADP水平发生扰动,这或许可以解释为什么二甲双胍在发挥其治疗益处的同时不会有明显的副作用[11]。
02抑制肝糖异生的分子机制
二甲双胍如何通过激活AMPK来降低血糖呢?众所周知,降低肝糖输出是二甲双胍最重要的降糖机制,其中又以抑制肝糖异生为主。二甲双胍可通过多种AMPK依赖途径来抑制糖异生:
二甲双胍通过肝脏AMPK途径抑制肝糖异生
二甲双胍可通过AMPK信号通路,激活非典型蛋白激酶C,进而引起CBP分子436丝氨酸磷酸化,导致CREB-CBP-CRTC2转录复合体解离,从而使下游糖异生基因的表达降低[12]。二甲双胍还可通过AMPK信号通路上调小异二聚体伴侣(SHP),SHP进而与转录因子CREB直接作用,阻止CREB对CRTC2的招募,从而下调糖异生基因的表达[13]。
二甲双胍通过肠道AMPK途径抑制肝糖异生
近年来研究发现,二甲双胍在被吸收前即可通过激活肠道的AMPK信号通路发挥抑制肝脏糖异生的作用[14]。经肠道给药后,二甲双胍迅速激活肠道AMPK及其下游信号通路,进而通过分布于肠道的迷走神经传入纤维,将局部信号传递至中枢,再通过迷走神经传出纤维支配肝脏,最终抑制肝脏的葡萄糖输出。
目前相关研究仍在不断深入,以期进一步阐明二甲双胍通过AMPK降糖的更多分子机制。
三、一靶多效:二甲双胍降糖外作用与AMPK机制密切相关
近年研究发现,二甲双胍的降糖外多效性作用也与AMPK机制密切相关,包括心肾保护、抗衰老、抗肿瘤以及神经系统保护等。
心血管保护:基础研究证实,在T2DM小鼠模型中,二甲双胍、运动单独或联合可激活AMPK并减少核转录因子-κB(NF-κB)介导的免疫反应,从而改善心肌纤维化[15]。另有研究表明,二甲双胍可能通过激活AMPK促进白色脂肪棕色化,发挥改善代谢异常和抗动脉粥样硬化的作用[16]。
肾脏保护:二甲双胍对糖尿病肾病有潜在保护作用,可以通过AMPK介导的信号通路缓解系膜细胞凋亡和足细胞损伤,保护肾脏免受氧化应激、炎症凋亡和上皮间质转化。二甲双胍可通过AMPK/SIRT1-FoxO1通路诱导肾脏自噬来减轻肾脏的损伤,改善氧化应激和糖代谢异常;还可通过AMPK-mTOR轴激活负性调节mTOR表达,增强自噬,从而缓解肾功能损伤[17]。
抗衰延寿:二甲双胍可通过激活AMPK信号,抑制下游相关分子,以此来实现延缓衰老、延长寿命的作用[18]。动物研究显示,二甲双胍与PEN2结合后,靶向激活溶酶体AMPK也是其抗衰老作用的重要通路[11]。
抑制肿瘤:二甲双胍激活AMPK后,可通过抑制雷帕霉素靶蛋白复合物1(mTORC1)的活性,促进肿瘤细胞凋亡[19];还可通过AMPK/PKA/GSK-3β通路,抑制survivin蛋白表达,并诱导其降解,促进肺癌细胞发生凋亡[20]。除了促进肿瘤细胞凋亡以外,二甲双胍还可通过激活AMPK通路,诱导肿瘤细胞周期停滞[19]。
神经系统保护:二甲双胍可以通过血脑屏障,作用于特定的神经元和神经胶质细胞,激活AMPK通路,调节神经能量代谢,减轻神经相关症状,保护外周神经和中央神经[21-23]。
四、结语
综上所述,AMPK与细胞生长、生存和多种代谢信号途径关系密切,且涉及糖尿病、肿瘤等多种疾病。尽管相关机制尚未完全阐明,但目前证据支持,二甲双胍之所以能发挥降糖及降糖外的多种“神奇”功效,很大一部分归因于其对AMPK的激活作用。图4总结了二甲双胍依赖于AMPK信号通路的细胞和分子机制[24]。对激活AMPK机制的深入挖掘,有助于临床更好地理解二甲双胍在糖尿病及其他疾病领域的临床获益,期待未来更多研究进一步揭示“神药”的秘密。

图4. 二甲双胍依赖于 AMPK信号通路的细胞和分子机制总览图
专家简介

匡洪宇 教授
哈尔滨医科大学附属第一医院内分泌科主任二级教授、博士研究生导师、首届龙江名医中华医学会糖尿病学分会常委兼视网膜病变学组组长中国医师学会内分泌代谢科医师分会常委中国健康管理学会糖尿病防治与管理专业委员会副主任委员中国研究型医院学会糖尿病学专业委员会副主任委员中国老年保健医学研究会老年健康教育分会副主任委员中国医学基金会内分泌专业委员会副主任委员兼副秘书长中国1型糖尿病联盟副主席黑龙江省医学会糖尿病学会主任委员黑龙江省糖尿病临床医学研究中心主任黑龙江省女医师协会会长黑龙江省内分泌代谢医师协会副主任委员黑龙江省内分泌学会副主任委员《中华糖尿病杂志》《中国糖尿病杂志》《国际内分泌代谢杂志》《Diabetes Care中文版》《Diabetologia中文版》《Lancet Diabetes & Endocrinology中文版》《Endocrine Reviews中文版》等杂志编委参考文献
1. 耿凤豪, 等. 心脏杂志. 2014; 26(1): 97-100.
2. Herzig S, Shaw RJ. Nat Rev Mol Cell Biol. 2018; 19(2): 121-135.
3. Afinanisa Q, et al. Int J Mol Sci. 2021; 22(20): 10921.
4. 袁虎, 等. 国际内分泌代谢杂志. 2010; 30(Suppl): 25-28.
5. Shaw RJ, et al. Science. 2012; 336(6083): 813-814.
6. Zhou G, et al. J Clin Invest. 2001; 108(8): 1167-1174.
7. Owen MR, et al. Biochem J. 2000; 348 Pt 3(Pt 3): 607-614.
8. Viollet B, et al. Clinical Science. 2012; 122(6): 253-270.
9. Larsen S, et al. Diabetologia. 2012; 55(2): 443-449.
10. He L, et al. Cell Metab. 2015; 21(2): 159-162.
11. Ma T, et al. Nature. 2022; 603(7899): 159-165.
12. He L, et al. Cell. 2009; 137(4): 635-646.
13. Kim YD, et al. Diabetes. 2008; 57(2): 306-314.
14. Duca FA, et al. Nat Med. 2015; 21(5): 506-511.
15. Liu J, et al. Biomed Pharmacother. 2023; 157: 114080.
16. Su M, et al. Clin Sci (Lond). 2020; 134(12): 1537-1553.
17. 邓煜璇, 等. 中国全科医学. 2024; 27(3): 262-267, 272.
18. 苗婷, 等. 医学信息. 2019; 32(14): 53-56.
19. 赵维, 等. 中国肿瘤生物治疗杂志. 2022; 29(4): 374-379.
20. Luo Z, et al. J Cell Biochem. 2019; 120(7): 11890-11899.
21. 肖建中. 中华糖尿病杂志. 2024; 16(7): 735-739.
22. Xu C, et al. Neuroreport. 2023; 34(3): 190-197.
23. Ismail TR, et al. Biology (Basel). 2023; 12(3): 480.
24. Hasanvand A. Inflammopharmacology. 2022; 30(3): 775-788.
