自2024年10月8日起,拟提交申请的医疗器械产品根据具体情况提交相应可用性注册申报资料,在审的医疗器械产品无需提交可用性注册申报资料。
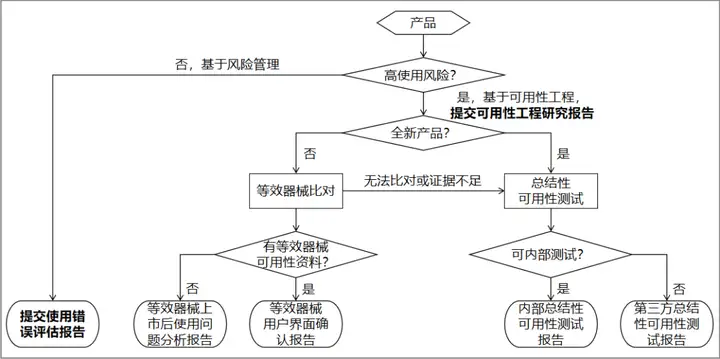
对于拟申请注册的医疗器械产品,高使用风险医疗器械提交可用性工程研究报告,中、低使用风险医疗器械若相应产品指导原则有可用性或可用性相关要求(如模拟使用等),则按其要求提交相应注册申报资料,其他情况均提交使用错误评估报告。
对于拟申请变更注册的医疗器械产品,无需补充变更前产品的可用性工程研究资料,若涉及用户、使用场景、用户界面的实质性更改则按前款要求提交可用性注册申报资料。
对于拟申请延续注册的医疗器械产品,原则上无需提交可用性注册申报资料。
3月19日,国家药监局器审中心已发布《医疗器械可用性工程注册审查指导原则》及应用说明,本期提供该文件的浓缩精华版,助力各位便捷读懂此指导原则。

本指导原则适用于第二类、第三类医疗器械可用性工程的注册申报,不适用于体外诊断试剂。
2.主要概念(一)可用性工程和可用性
可用性工程是指综合运用关于人类的解剖、生理、心理、行为、文化等方面能力与限制的知识来设计开发医疗器械,以增强医疗器械的可用性。
可用性(Usability)是指预期用户在预期使用场景下正常使用医疗器械时,保证医疗器械安全有效易于使用的用户界面特性,包括但不限于易读性、易理解性、易学习性、易记忆性、易操作性、用户差错防御性等特性。
(二)用户、使用场景和用户界面 :可用性工程的三个核心要素。
1.用户:指注册申请人所规定的与医疗器械交互的全部人员,如医务、患者、家庭护理、清洁、运输、安装、维护、维修、处置等人员。
2.使用场景
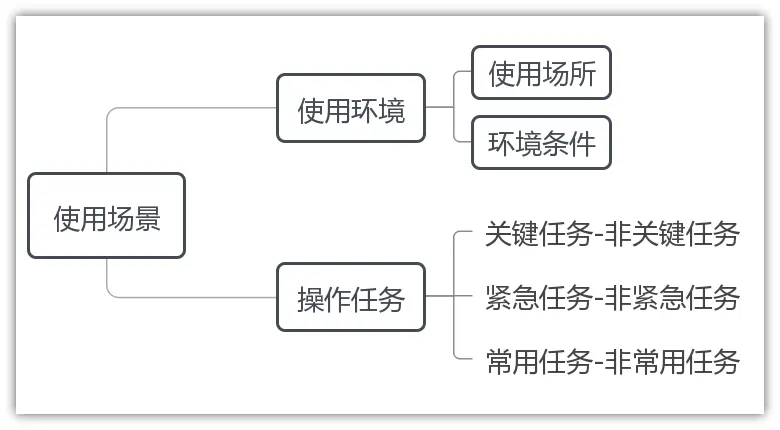
3.用户界面:即用户接口,指用户与医疗器械人机交互的全部对象及方式,包括但不限于医疗器械的形状、尺寸、重量、显示、反馈、连接、组装、操作、控制、说明书、标签、包装、用户培训材料等。
(三)医疗器械使用情况和用户操作情形
1.医疗器械使用情况

2.用户操作情形

(一)可用性工程定位
(二)使用风险导向
(三)全生命周期管理
本指导原则基本思路如图所示↓
医疗器械设计开发的重要组成部分,注册申请人需在质量管理体系设计开发过程的框架下建立充分、适宜、有效的可用性工程过程,包括用户界面的需求分析、设计、实现、验证、确认、更改等活动,风险管理和可追溯性分析贯穿于其中,且每个活动均需形成相应可用性工程文档。
5.用户界面验证与确认用户界面验证又称形成性评价,用户界面确认又称总结性评价,用户界面验证是用户界面确认的基础。
6.技术考量(一)临床试验
(二)进口医疗器械:原则上需基于使用风险级别在中国开展用户界面确认工作,除非提供数据详实的支持材料证实中外差异对于用户界面确认无显著影响。
(三)现成用户界面:注册申请人未进行(含无法证明)完整可用性工程生命周期控制的用户界面。
(四)组合使用
(五)标准:可根据可用性工程、人因工程、人类工效学、人体工程学、职业安全相关国际、国家和行业标准开展医疗器械可用性工程工作。
(六)可用性工程更改
7.可用性工程研究资料(一)可用性工程研究报告
1.基本信息
2.使用风险级别
3.核心要素:明确用户、使用场景、用户界面。
4.可用性工程过程
5.用户界面需求规范
6.使用风险管理
7.用户界面验证与确认
8.用户界面可追溯性分析
9.用户培训方案
10.结论:可用性工程过程和结果,说明综合剩余使用风险是否均已降至可接受水平,判定用户界面安全有效性是否满足要求。
(二)使用错误评估报告
1.基本信息:明确名称、型号规格、预期用途、适用人群、结构组成。
2.使用风险级别
3.核心要素:明确用户、使用场景、用户界面。
4.同类医疗器械上市后使用问题分析
5.使用风险管理
6.结论:简述申报医疗器械使用错误评估结果,说明综合剩余使用风险是否均已降至可接受水平,判定用户界面安全有效性是否满足要求。
8.注册申报资料补充说明(一)产品注册
1.研究资料
2.说明书与标签
(二)变更注册:根据可用性工程更改情况,在“CH3.5.11可用性/人为因素”提交相应变化对于产品安全性与有效性影响的研究资料。
(三)延续注册:通常无需提交可用性工程研究资料。若适用,根据注册证“备注”所载明的要求提交相应可用性工程研究资料。
【内容及图片来源:国家药监局器审中心网站】
