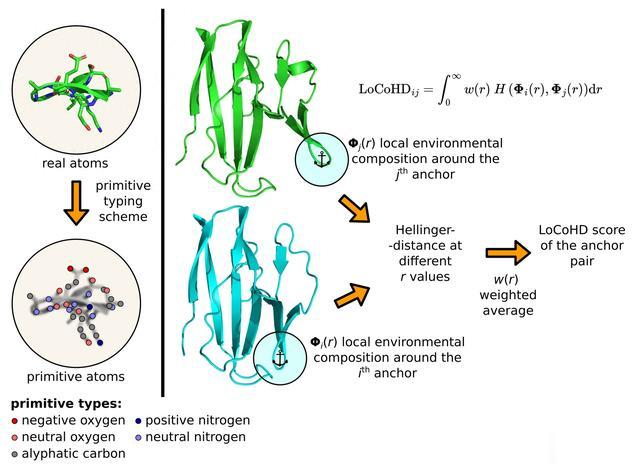
该图显示了为给定的一对锚计算LoCoHD分数的逐步过程
在最近发表在《自然通讯》上的一篇论文中,hunn - ren - elte蛋白质建模研究小组(化学研究所)为一种数学方法奠定了基础,该方法允许计算机辅助比较蛋白质的三维结构。这种方法的独特之处在于,目前可用的替代方法只考虑了原子的位置,而这种名为LoCoHD(局部组成海灵格距离)的新技术还包括了原子的化学信息。
蛋白质是分子机器,执行细胞功能所需的过程,充当分子开关,转录DNA信息,运输小分子和大分子,调节代谢相关的化学反应。然而,要使这一切成功,所讨论的蛋白质必须具有正确的空间构象,即其自身正确的3D排列。
有几种实验方法(x射线晶体学、核磁共振波谱学、低温电子显微镜)可用于确定蛋白质中原子的排列,在过去的几十年里,蛋白质研究人员已经发现了近22万种蛋白质的形状。这些结果越来越需要能够分析这些排列的计算方法的发展。
其中一种方法是名为LoCoHD的算法,它是由ELTE Hevesy György化学学院的博士候选人、András Perczel博士研究小组的研究员Zsolt Fazekas开发的。该算法根据蛋白质的化学性质(例如,元素组成、电荷、疏水性等)来比较氨基酸周围的局部环境。
该方法以0到1的简单比例决定所讨论的结构彼此之间的差异。接近0的值表明原子排列和化学性质之间高度相似,而接近1的值表明所比较的蛋白质可能具有非常不同的性质。由此产生的数值(所谓的度量)可以用来获得关于所研究系统的新信息。
该算法使用多步协议生成表示结构差异的数字。在第一步中,它将蛋白质中的真实原子转化为所谓的原始原子。这些可以表示为虚拟标记的位置,其标记告诉原始原子的化学性质。

图中显示了两种强双峰氨基酸——足蛋白中His276和Met197的LoCoHD曲线(上图)和结构(下图)
例如,一个原始原子可以是“带正电的氮”、“带负电的氧”、“带中性电的氧”、“芳香碳”等等。标签是根据所谓的原始类型模式生成的,该模式以表格的方式告诉我们如何将真实原子转换为原始原子。用户可自由指定此表,固定化学分辨率的方法。
第二步是通过选择基本原子的子集来确定比较的参考点。这些选定的特殊原始原子称为锚原子。对于选择的每个锚原子对,算法执行一个比较步骤,比较结果给出我们想要的不相似度度量。这些数字可以在局部水平上使用,或者它们可以被平均成一个描述整个蛋白质的描述符。
在这项研究中,研究人员强调,该方法也可以用于两年一次的CASP (Critical Assessment of Protein Structure Prediction,蛋白质结构预测关键评估)竞赛,这是蛋白质研究领域的一个知名竞赛。在这次比赛中,参赛者使用不同的算法来模拟尚未公布结构的蛋白质的形状。CASP的评委使用许多结构比较方法来评估竞争者,但这些方法都没有考虑到当地氨基酸环境的化学性质。
利用2020年CASP14竞赛的数据,研究人员现在对几种模型蛋白质进行了比较分析,包括基于人工智能的AlphaFold2方法预测的结构。其中,他们强调了对SARS-CoV-2病毒中一种名为ORF8的蛋白质的分析。在这种蛋白质的模型结构中,氨基酸环境在相互作用模式上与实验结构中发现的环境有很大的不同。
除了研究静态结构外,研究人员还测试了该方法是否适用于分析蛋白质的内部运动。他们使用了能够再现分子运动的模拟和从结构集合中提取的数据。正在研究的系统之一是足蛋白,它在肾脏中起着至关重要的作用,其突变可能导致严重的,通常是致命的疾病。
LoCoHD方法用于鉴定蛋白质中在podocin运动过程中发生重大化学环境变化的氨基酸,这些变化会影响其结构和功能。
