
引言
蛋白质与 DNA 的结合特异性预测是一项复杂但至关重要的任务,对于理解基因调控具有重要意义。通常情况下,蛋白质-DNA复合物会与特定的DNA靶位点结合,而蛋白质会以不同的结合特异性与多种DNA序列结合。为了捕捉这种信息,8月5日Nature Methods的研究报道“Geometric deep learning of protein–DNA binding specificity”,开发了结合特异性深度预测器(DeepPBS),这是一种几何深度学习模型,旨在从蛋白质-DNA结构中预测结合特异性。DeepPBS可以应用于实验或预测结构,提取残基的可解释蛋白质原子重要性评分,并通过突变实验验证这些评分。转录因子在各种调控功能中起着关键作用,因此,了解蛋白质如何靶向特定DNA序列的机制至关重要。研究发现,结合机制包括精确的静电相互作用、脱氧核糖-苯丙氨酸堆叠、鸟嘌呤和精氨酸在大沟中的双齿氢键等。蛋白质-DNA结构通常通过X射线晶体学、核磁共振光谱或冷冻电子显微镜实验获得,并存储在蛋白质数据银行(PDB)中。尽管这些结构显示了一个结合的DNA序列和相关的物理化学相互作用,但它们并不包含所有可能结合的DNA序列。相反,结合特异性数据可以通过蛋白质结合微阵列、SELEX 测序、染色质免疫沉淀测序等实验获得,但这些实验不提供结构信息。因此,需要手动检查以将结构数据中的分子相互作用细节与结合特异性数据相关联。预测特定蛋白质序列的结合特异性仍然是一个具有挑战性的未解决问题。蛋白质-DNA结构包含有价值的信息,人工智能可以利用这些信息在蛋白质家族中实现广泛的通用性。在这个框架下,研究人员开发了 DeepPBS,它是蛋白质家族无关的,可以处理生物组装体,并预测DNA 序列偏好。DeepPBS的输入不限于实验结构。蛋白质结构预测方法(如 AlphaFold、OpenFold 和 RoseTTAFold)以及蛋白质-DNA复合体模型(如RoseTTAFoldNA、RoseTTAFold All-Atom、MELD-DNA和 AlphaFold3)的快速发展,导致可用于分析的结构数据呈指数增长。这种情况突显了需要一个通用的计算模型来分析蛋白质-DNA结构。该研究展示了DeepPBS如何与结构预测方法结合使用,以预测没有实验结构的蛋白质的特异性。通过优化结合DNA的DeepPBS反馈,可以改进蛋白质-DNA 复合物的设计。DeepPBS 提供了一种基础,用于机器辅助研究,进而推动我们对分子相互作用的理解,并指导实验设计和合成生物学研究。 转录因子在各种调控功能中起着关键作用,因此,了解蛋白质如何靶向特定 DNA 序列的机制至关重要。蛋白质-DNA结构通常通过X射线晶体学、核磁共振光谱或冷冻电子显微镜实验获得,并存储在蛋白质数据银行(PDB)中。尽管这些结构显示了一个结合的 DNA 序列和相关的物理化学相互作用,但它们并不包含所有可能结合的 DNA 序列 。相反,结合特异性数据可以通过蛋白质结合微阵列、SELEX 测序、染色质免疫沉淀测序等实验获得,但这些实验不提供结构信息。因此,需要手动检查以将结构数据中的分子相互作用细节与结合特异性数据相关联。为了应对这一挑战,研究人员开发了 DeepPBS 模型,其输入不限于实验结构,还可以使用预测结构。DeepPBS 框架DeepPBS 处理蛋白质和 DNA 组成的结构,作为一个双分图,分别对蛋白质和 DNA 成分进行独特的空间图表示。蛋白质图是一个基于原子的图,重原子作为顶点。计算了这些顶点上的多个特征。将 DNA 表示为对称螺旋(sym-helix),这种表示法去除了 DNA 所具有的任何序列标识,同时保留了双螺旋的形状。可以选择将 DNA 序列信息重新引入 sym-helix 点上作为特征。DeepPBS 在蛋白质图上执行一系列空间图卷积,以聚合原子邻域信息。接下来,DeepPBS 的关键组件包括从蛋白质图到 sym-helix 的一组双分图几何卷积。具体的化学相互作用(例如氢键)依赖于位置和方向。DeepPBS 学习 sym-helix 点的几何方向与邻近蛋白质残基的方向和化学性质之间的关联。四种不同的双分图卷积用于 sym-helix 点,分别对应于大沟、小沟和磷酸基团和糖基团。大沟和小沟卷积被称为 '沟读取'。磷酸基和糖基卷积,结合 DNA 形状信息,形成 '形状读取'。'沟读取' 和 '形状读取' 因子协同作用,不同程度地决定了不同蛋白质家族的结合特异性 。
转录因子在各种调控功能中起着关键作用,因此,了解蛋白质如何靶向特定 DNA 序列的机制至关重要。蛋白质-DNA结构通常通过X射线晶体学、核磁共振光谱或冷冻电子显微镜实验获得,并存储在蛋白质数据银行(PDB)中。尽管这些结构显示了一个结合的 DNA 序列和相关的物理化学相互作用,但它们并不包含所有可能结合的 DNA 序列 。相反,结合特异性数据可以通过蛋白质结合微阵列、SELEX 测序、染色质免疫沉淀测序等实验获得,但这些实验不提供结构信息。因此,需要手动检查以将结构数据中的分子相互作用细节与结合特异性数据相关联。为了应对这一挑战,研究人员开发了 DeepPBS 模型,其输入不限于实验结构,还可以使用预测结构。DeepPBS 框架DeepPBS 处理蛋白质和 DNA 组成的结构,作为一个双分图,分别对蛋白质和 DNA 成分进行独特的空间图表示。蛋白质图是一个基于原子的图,重原子作为顶点。计算了这些顶点上的多个特征。将 DNA 表示为对称螺旋(sym-helix),这种表示法去除了 DNA 所具有的任何序列标识,同时保留了双螺旋的形状。可以选择将 DNA 序列信息重新引入 sym-helix 点上作为特征。DeepPBS 在蛋白质图上执行一系列空间图卷积,以聚合原子邻域信息。接下来,DeepPBS 的关键组件包括从蛋白质图到 sym-helix 的一组双分图几何卷积。具体的化学相互作用(例如氢键)依赖于位置和方向。DeepPBS 学习 sym-helix 点的几何方向与邻近蛋白质残基的方向和化学性质之间的关联。四种不同的双分图卷积用于 sym-helix 点,分别对应于大沟、小沟和磷酸基团和糖基团。大沟和小沟卷积被称为 '沟读取'。磷酸基和糖基卷积,结合 DNA 形状信息,形成 '形状读取'。'沟读取' 和 '形状读取' 因子协同作用,不同程度地决定了不同蛋白质家族的结合特异性 。
DeepPBS框架的示意图(Credit: Nature Methods)
输入数据: 图a 显示了DeepPBS的输入数据来源,包括实验数据(如PDB中的蛋白质- DNA 复合物结构)、分子模拟快照以及设计的复合物。图中以PDB ID 2R5Y为例,展示了输入数据的形式。蛋白质结构表示: 图b 展示了蛋白质结构的图表示方法。重原子作为顶点,每个顶点计算了一些特征(例如原子类型、电荷、半径等)。这些特征作为图卷积的输入。DNA 结构表示: 图c 描述了对称螺旋(sym-helix)在DNA结构中的应用,通过在碱基对框架中应用对称模式,将DNA结构转化为对称螺旋,保留双螺旋形状,但去除了序列特异性。图卷积操作: 图d 解释了在蛋白质图上的空间图卷积,用于聚合原子邻域信息。随后,将双分图几何卷积应用于蛋白质图顶点到sym-helix点(以不同颜色的球体表示大沟、小沟、磷酸和糖基)。三维到一维转换: 图e 显示了将聚合信息从三维对称螺旋转化为一维表示的方法,包括形状特征。随后进行一维卷积和回归,以预测碱基对概率。输出结果: 图f 展示了DeepPBS的输出,即结合特异性的预测结果。可解释性分析: 图g 说明了通过扰动双分图边缘,评估其对输出结果变化的影响,从而提供有效的可解释性度量。DeepPBS 的训练与验证为了训练和验证 DeepPBS,研究人员构建了一个包含 523 个数据点的交叉验证集,每个数据点对应一个生物组装体,包含一个蛋白质链和一个相应的 PWM。使用非对齐的局部对齐过程将 PWM 与结构中的 DNA 对齐,以创建用于损失/度量计算的对应关系。对于每个折叠,使用剩余四个折叠训练的模型进行交叉验证预测。此外,还创建了一个基准数据集(对应于 130 个蛋白质链的生物组装体)用于评估 DeepPBS 的性能。该数据集的采样遵循与交叉验证集相同的质量标准,每个簇最多采样五个成员。模型性能的评估DeepPBS 的架构允许模型通过 '沟读取' 和 '形状读取' 两种机制进行训练。实验结果显示,DeepPBS 框架对结合特异性的预测优于单独使用 '沟读取' 或 '形状读取' 的模型。此外,整合序列信息到 sym-helix 点中('DeepPBS with DNA SeqInfo')可以提高性能,显著缩小数据集中的固有性能差距 。实验结构中的性能DeepPBS 使用了一组基准测试集来评估模型的性能。结果显示,DeepPBS 框架允许模型通过 '沟读取' 和 '形状读取' 两种机制进行训练,实验结果显示 DeepPBS 框架对结合特异性的预测优于单独使用 '沟读取' 或 '形状读取' 的模型 。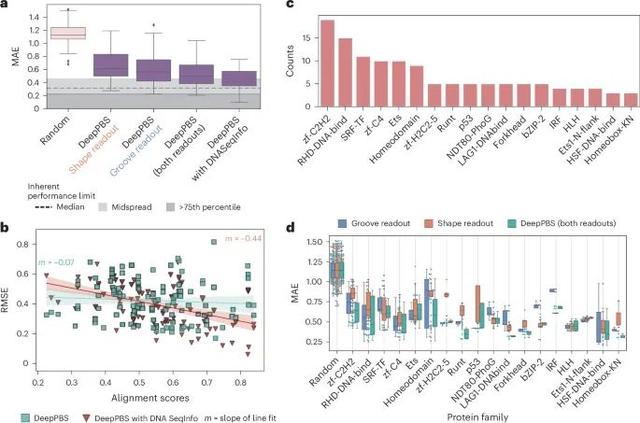
DeepPBS在预测实验确定结构的蛋白质家族结合特异性中的表现(Credit: Nature Methods)
DeepPBS模型及其变体的预测性能: 图a 显示了DeepPBS模型及其变体(包括“沟读取”、“形状读取”和“带DNA序列信息的DeepPBS”)在基准测试集上的预测性能。图中展示了平均绝对误差(MAE)和均方根误差(RMSE)的箱线图。结果显示,整合了DNA序列信息的DeepPBS模型表现最佳,其次是综合了“沟读取”和“形状读取”的DeepPBS模型。与共晶衍生DNA比对得分的关系: 图b 展示了DeepPBS和“带DNA序列信息的DeepPBS”模型在PWM(位置权重矩阵)与共晶衍生DNA比对得分方面的表现。结果表明,带DNA序列信息的DeepPBS模型的线性拟合斜率较高,而DeepPBS模型的斜率接近于零,表明DeepPBS模型对DNA序列的敏感性较低。蛋白质家族的分布: 图c 描述了基准测试集中不同蛋白质家族的丰度(基于PFAM注释)。该图显示了基准测试集中各个蛋白质家族的成员数量,涵盖了多种DNA结合蛋白家族。不同蛋白质家族的模型表现: 图d 展示了DeepPBS、“沟读取”和“形状读取”模型在各种蛋白质家族中的表现(仅包括成员数量大于3的家族)。结果表明,DeepPBS模型在大多数蛋白质家族中表现出色,尤其是在家族成员较少的情况下(如热休克因子蛋白家族)。蛋白质家族特定结合模式DeepPBS 框架捕捉到了蛋白质家族特定的结合模式,通过分析 p53- DNA 接口中的残基重要性,研究人员验证了 DeepPBS 的预测结果与实验数据的一致性 。模拟预测的蛋白质-DNA复合体的应用DeepPBS 还应用于合成设计的蛋白质-DNA 复合体,预测了特定 DNA 序列的结合特异性,并与实验结果进行比较,验证了 DeepPBS 的预测准确性 。蛋白质残基在 p53-DNA 接口处的重要性评估DeepPBS 的架构允许有意激活或停用双分图几何卷积阶段的特定边缘,使用原始和改变的预测结果之间的均方差来量化扰动边缘集在确定结合特异性中的影响 。
通过p53-DNA接口的DeepPBS重要性评分可视化作为案例研究,并进行实验验证(Credit: Nature Methods)
p53与DNA结合界面的重原子重要性评分: 图a 展示了DeepPBS计算的p53蛋白与DNA结合界面中重原子的相对重要性(RI)评分,这些评分通过对5 Å范围内的重原子进行标准化处理得到。图中以球体大小表示重原子的RI评分,评分范围从0到1不等。特定残基与DNA的相互作用细节: 图b 到图e 是对特定残基(Lys120B、Arg280A、Cys277A 和 Arg248B)与DNA相互作用的放大视图,展示了这些残基的重要性评分和相应的化学相互作用。例如,Lys120B与DNA大沟中的G形成氢键,以及与DNA骨架磷酸形成氢键。残基重要性评分的聚合: 图f 显示了通过平均和最大聚合方法计算的残基重要性评分(前20名残基)。这些评分反映了各个残基在蛋白质-DNA结合中的重要性。DeepPBS的结合特异性预测: 图g 展示了DeepPBS对p53-DNA结合特异性的预测结果。该预测结果与已知的p53结合模式(RRRC(A/T)(A/T)GYYY)一致。残基重要性评分与实验自由能变化(ΔΔG)的比较: 图h 是DeepPBS预测的残基重要性评分与通过丙氨酸扫描突变实验测定的自由能变化(ΔΔG)之间的回归图和Pearson相关系数(PCC)。结果显示,两个数据集之间的PCC为0.605,表明DeepPBS的重要性评分与实验数据之间存在显著相关性。DeepPBS 提供了一种基础,用于机器辅助研究,进而推动对分子相互作用的理解,并指导实验设计和合成生物学研究。DeepPBS 的当前版本有固有的局限性,主要适用于双链 DNA,尚未应用于单链 DNA、RNA 或化学修饰碱基。然而,随着数据的增加和模型的改进,DeepPBS 具有扩展到其他聚合物- 聚合物相互作用和机制突变的潜力 。总结来说,该研究引入了一个计算框架,该框架提炼了蛋白质- DNA 结合的复杂结构细节,并将这些理解与结合特异性数据联系起来,有效地连接了结构确定和特异性确定实验。DeepPBS 架构可用于评估家族特定的 '沟读取' 和 '形状读取' 模式及其对结合特异性的影响。尽管结构预测方法如 RFNA 和 AlphaFold3 可以预测从给定的蛋白质和 DNA 序列到复合体,但它们无法提供结合特异性的见解。DeepPBS 在预测蛋白质-DNA 复合体的建模