一、FDA监管关注点
不少医疗器械相关事件和事故往往涉及:设计缺陷、使用错误、不符合用户需求等问题,这对患者安全和健康构成严重威胁,FDA关注到前述问题与人因工程的关联性,即:用户与医疗器械之间的交互和界面设计。

二、FDA重要监管事件

三、FDA适用法规、指南、标准
法 规:
医疗器械制造商应当符合质量体系法规21 CFR Part820,尤其是第30节设计控制,其中包括人因工程相关要求。
指 南:
·2016年发布的《Applying Human Factors and Usability Engineering to Medical Devices》第2版指南文件。
·人因审核优先级最高的器械清单Listof Highest Priorities Device for Human Factor Review
·医疗器械软件上市前递交指南Guidance for the Content of Premarket Submissions for Software Contained in MedicalDevices
·指南-产品整个生命周期:输液泵上市前通知[510(k)]递交Guidance for Industry and FDA Staff - Total Product Life Cycle:Infusion Pump - Premarket Notification [510(k)] Submissions
·医疗机构中的再加工医疗器械:验证方法和标识Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling
·医疗器械患者标识指南Guidanceon Medical Device Patient Labeling; Final Guidance for Industry and FDA Reviewers
·器械建议的标识要求Labeling requirements from Device Advice
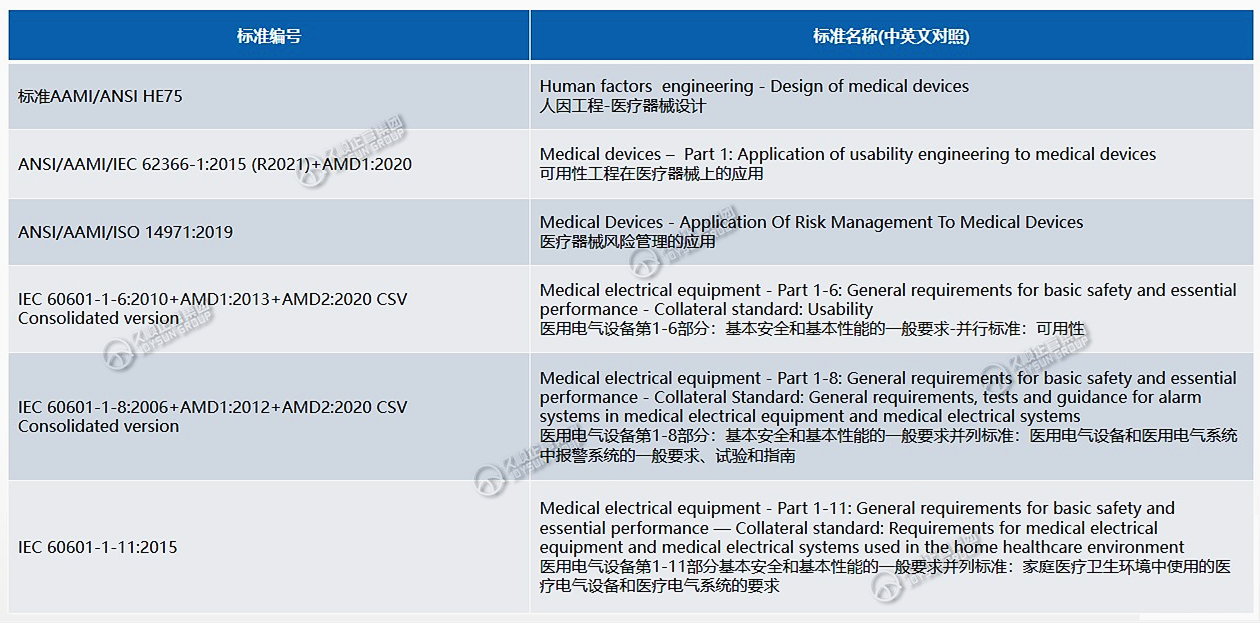
FDA认可共识标准:
四、FDA审核主体与要求
1.FDA的CDRH及其它中心对各种上市前申请进行审核,人因上市前评估组(Human Factors Premarket Evaluation Team)是审核申请资料团队的顾问。该评估组可对器械制造商提出的人因评估和测试方式的问题给出解答,可通过与制造商进行远程会议或面对面会议的方式,与制造商共同解决上市前申请中的人因缺陷问题。
2.上市前申请中的人因工程或可用性工程(HFE/UE)报告应当提供总结,内容是关于器械使用安全性和有效性的信息。
3.该报告应当讨论与安全相关的人因工程或可用性工程的考量、问题、过程、解决方式、结论等。
4.递交资料应当充分描述器械所有严重使用风险的识别、评估、最终评价。
5.为便于FDA审核,在人因工程或可用性工程中直接使用的材料(包括:用户与器械交互风险分析和具体风险分析过程的结果和结论),应当体现于人因工程或可用性工程报告中。
五、报告结构建议
1.结 论
2.预期用户、预期用途、预期使用环境和培训的描述
3.用户交互的描述
4.已知使用问题的总结
5.器械使用相关危险源和风险的分析
6.初步分析和评估的总结
7.关键任务的描述和分类
8.人因工程确认测试的细节
