
在循环过程中,富锂氧化物阴极由于原子无序和纳米结构重排而导致能量密度降低,但它们难以表征。近日,牛津大学M. Saiful Islam、巴斯大学Kit McColl等人使用从头算分子动力学和基于团簇扩展的蒙特卡罗模拟组合方法,在层状富锂(Li1.2–xMn0.8O2)阴极中研究了该过程的动力学和热力学。
计算方法
作者使用VASP进行GGA + U AIMD模拟,并使用Perdew–Burke–Ernzerhof+U泛函来处理电子交换关联作用,其中Mn 3d轨道的Hubbard U参数为3.9 eV,以及使用Grimme的D3方法进行色散校正。此外,作者设置了400 eV的平面波截断能,并采用单个Γ点的k点网格进行布里渊区采样,以及时间步长为2 fs。
为了对Li0.2Mn0.8O2中的结构转化途径进行采样,作者在300、600和900 K下进行了AIMD模拟,并且从脱锂框架开始,使用(1 × 2 × 1)的带状超胞结构(160个原子)。对于每个AIMD模拟,作者首先在2 ps内利用微正则(NVE)系综将温度从0 K上升至目标温度,其中每50步调整一次温度。然后,在目标温度下利用正则(NVT)系综进行2 ps的AIMD模拟。
结果与讨论

如图1a所示,富锂O2-Li1.2Mn0.8O2晶体结构含有八面体配位Li和Mn的O2-堆叠层,氧离子与三个(O–Mn3)或两个(O-Mn2)Mn原子配位,其中O–Mn2原子与富Mn层中的Li离子配位。为了理解O2-Li1.2Mn0.8O2在O-氧化还原状态下的第一次循环性质,作者通过每化学式单位去除一个锂离子的方法得到了Li0.2Mn0.8O2结构。作者通过DFT驰豫发现,这种脱锂结构在结构重排中保持稳定,并且没有Mn重排或氧二聚化(图1b)。

如图2a所示,O–Mn2位点中的O离子优先被氧化,然后层间过氧化物(O22-)和超氧化物(O2-)物种从相邻过渡金属层中的O–Mn2位点形成(图2b,c,结构II)。一旦这些O原子发生二聚,它们与相邻Mn离子的键合相互作用就会减少,从而驱动Mn迁移到层间,并且跳跃到第一或第二相邻位点。这种Mn迁移导致O–O二聚体与Mn脱配,形成一对O2分子(结构III)。在更长的时间尺度上(约400 ps),六个Mn离子迁移到层间,并且形成更多的O2分子,它们聚集在一起形成Mn空位簇(结构IV)。该过程在动力学上是可行的,并保持系统的整体热力学稳定,以及结构IV比起始结构I更稳定(图2d)。
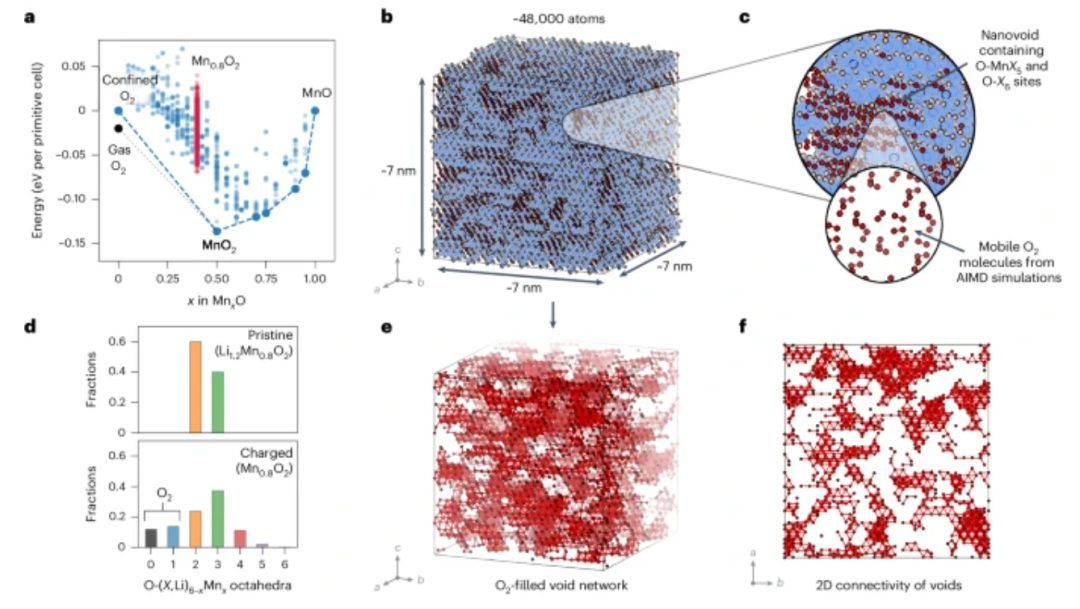
为了确定在多次循环后阴极(Mn0.8O2)的热力学稳定结构,并考虑无序和纳米级结构变化,作者开发了O-氧化还原的团簇扩展模型。对于该团簇扩展模型,作者使用混合DFT计算了沿O2–MnO2–MnO路线的结构能量,并考虑了自由气态O2和限制在本体中的O2分子。如图3a所示,Mn0.8O2结构在O2态和MnO2的基态之上,这表明Mn0.8O2为亚稳态。气态O2态低于受限O2态,表明O2从系统中损失。此外,对于限制在本体中的O2分子,Mn0.8O2也高于基态。因此,Mn0.8O2在热力学上易于分解为MnO2和O2,即使O2分子被限制在本体中并且不能从系统中损失。
为了搜索低能结构并分析纳米级特征,作者使用团簇扩展模型对包含48000个原子的结构进行晶格蒙特卡罗退火模拟。如图3b所示,作者模拟预测了局部相偏析行为,其中形成了含有受限O2分子的富含MnO2区域和缺乏Mn的纳米空隙(图3c)。系统中大约20%的O原子形成O2分子,而在表面形成的O2分子将快速损失。然而,如果在本体中形成的O2分子不能逃离,它们将被系统捕获。此外,相偏析也会导致无序(图3d)和层状结构的损失。O2填充纳米空隙的大小在0.5 nm至>1.5 nm之间变化,并且形成连接90%至95%的O2分子三维(3D)渗滤网络。这种渗滤网络允许O2通过阴极传输,但其高度曲折(图3e、f)。

如图4a所示,Mn和晶格O2–对的径向分布函数(RDF)具有尖锐的峰值,表明存在有序的结晶结构。相比之下,Mn–O2 RDF具有更宽的峰,峰之间具有非零值,表明分子O2是可移动的。如图4b所示,阴极体O2在比液体O2小得多的距离处具有第二相邻峰值最大值,并且具有大约1.45 g cm–3的密度。该密度高于液态O2的密度,但低于固体β-O2的密度,因此阴极体中的O2处于超临界状态。为了分析室温O2传输,作者在含有六个O2分子的渗透Mn空隙路径的Li0.2Mn0.8O2系统上进行了AIMD模拟(图4d–f)。晶格O2–离子的MSD几乎没有变化(图4c),表明O2–离子没有发生扩散。相反,O2分子和Li+离子的MSD随着时间的推移而增加,表明这两种物质发生显著的扩散,并且O2分子和Li+离子的扩散系数为1 × 10–7 cm2 s–1和~7 × 10–8 cm2 s–1。

如图5所示,通过DFT结构弛豫预测带状Li0.2Mn0.8O2超胞结构是亚稳态的,并且在实验时间尺度上发生重排。这些结构重排的动力学和热力学可以分别使用AIMD和团簇扩展蒙特卡罗进行模拟,并且用AIMD和混合DFT弛豫模拟动力学过程,从而确定了由层间O–O二聚引发的O-氧化还原机制,以形成稳定的O2分子。使用团簇扩展模型和蒙特卡罗模拟的热力学模拟确定了含有O2的纳米级空隙形成,AIMD将纳米空隙中的O2表征为高密度纳米受限流体。
结论与展望
作者发现了一种在本体中形成O2分子的动力学和热力学机制,其涉及Mn迁移并由层间氧二聚化驱动。体结构相分离为富含MnO2的区域和缺乏Mn的纳米空隙,其中包含作为纳米受限流体的O2分子。此外,这些纳米空隙以渗滤网络连接,并能实现长程氧传输,从而将本体O2的形成与表面O2的损失联系起来。该研究突出了开发动态稳定富锂O-氧化还原阴极本体结构策略的重要性,从而保持其高能量密度。
文献信息
Kit McColl et.al. Phase segregation and nanoconfined fluid O2 in a lithium-rich oxide cathode,Nature Materials,2024 https://doi.org/10.1038/s41563-024-01873-5


