随着高通量技术的发展,宏基因组学已经成为揭示研究微生物群落多样性及功能的前沿科学,通过宏基因组测序手段,可以发现难培养或不可培养微生物中的天然产物以及处于“沉默”状态的天然产物,在微生态学、海洋微生物资源开发、环境保护和污染修复、医学领域以及生物酶制剂开发等研究领域具有广泛应用。然而,宏基因组的数据处理给研究人员带来了巨大的挑战。近日,由Jim Shaw 和Yun William Yu在《Nature Biotechnology》上发表题为“Rapid species-level metagenome profiling and containment estimation with sylph”的文章,该文章介绍了Sylph这一新工具,基于零膨胀泊松(zero-inflated Poisson)k-mer统计,提供了精确的物种水平宏基因组分析。

通讯作者: Jim Shaw 和Yun William Yu
发表题目: Rapid species-level metagenome profiling and containment estimation with sylph
发表期刊: Nature Biotechnology
影响因子:33.1
发表时间:2024-10-08
通过对数据库进行宏基因组分析,可以检测和定量微生物,即使是在不可能组装的低丰度。我们引入了Sylph,一个物种水平的宏基因组profler,通过零感染泊松k-mer统计量估计基因组到宏基因组包含的平均核苷酸同一性(ANI),使基于ANI的类群检测成为可能。在宏基因组CAMI2海洋数据集上,Sylph是7个测试中最准确的剖面方法。对于多样本分析,与Kraken2相比,Sylph的运行时间快了10倍,并减少了30倍的内存,工作效率大大提高。Sylph的ANI估算值提供了一个与丰度正交的信号,该方法在289,232个基因组中证实了丁酸盐与帕金森病(PD)之间的关联,同时在菌株水平证实了已知的丁酸盐- PD关联。Sylph在不到1分钟和16 GB的随机访问内存中对85205个原核生物和2,917,516个病毒基因组进行宏基因组分析,与RefSeq相比,在人类肠道中检测出的病毒序列多30倍。即使对于低覆盖度的基因组,Sylph也能提供精确、高效的分析方法。
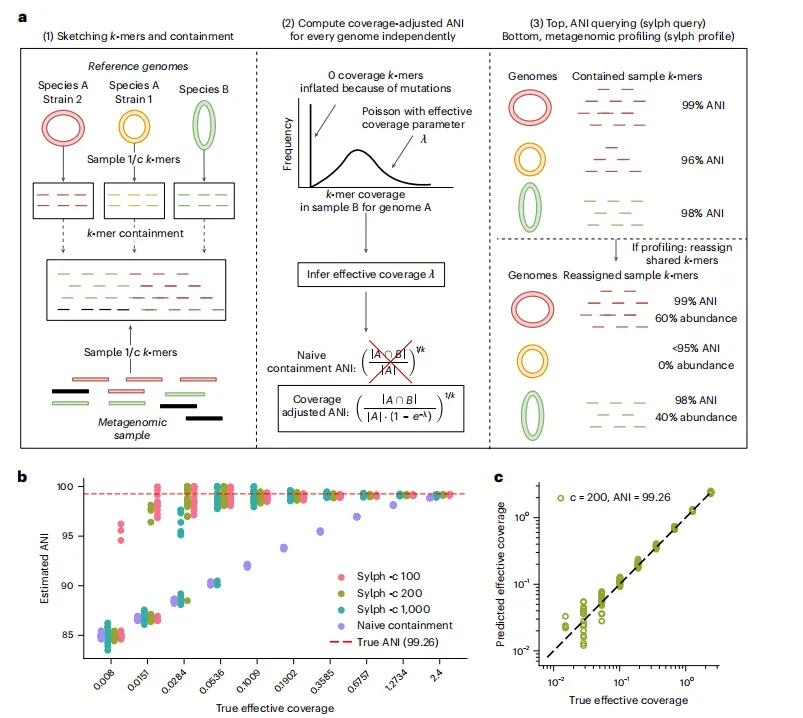
图1 sylph的算法概述及其在一次分离测序运行中的演示
文献链接
https://doi.org/10.1038/s41587-024-02412-y
