作者:张梦汀 郭梁洁 高越 王溦 姜锦成 王红丹
第一作者单位:郑州大学人民医院 河南省人民医院医学遗传研究所
通信作者:王红丹,郑州大学人民医院 河南省人民医院医学遗传研究所
引用本文: 张梦汀, 郭梁洁, 高越, 等. DNAH5新发剪接位点变异导致原发性纤毛运动障碍的遗传学分析[J]. 中华结核和呼吸杂志, 2025, 48(4): 365-372. DOI: 10.3760/cma.j.cn112147-20241011-00597.

摘要
目的探讨动力蛋白轴突重链5(DNAH5)基因内含子区变异对该基因转录剪接的影响。回顾性分析中国人群中DNAH5变异导致原发性纤毛运动障碍患者的表型特征。方法以家系中两例反复肺部感染、支气管扩张等呼吸系统症状就诊的患者为研究对象,对家系成员进行全外显子组测序查找可能的遗传学病因,Sanger测序对候选变异进行验证。利用Minigene剪接变异体分析技术研究新发现的内含子区变异对基因剪接的影响。通过文献检索和筛选,总结DNAH5不同基因突变类型导致原发性纤毛运动障碍患者的表型特征。结果两例患者同时携带父源性的DNAH5基因c.12367C>T(p.His4123Tyr)和c.1731-18A>G变异以及母源性的c.1933C>T(p.Gln645*)变异。参照美国医学遗传学与基因组学学会相关指南,DNAH5基因c.1933C>T(p.Gln645*)变异被判定为致病性变异(PVSl+PM2_Supporting+PP4),c.1731-18A>G变异被判定为致病性变异(PVSl+PM2_Supporting+PM3_Supporting+PP4),c.12367C>T变异被判定为可能良性变异。中国人群中DNAH5突变导致的患者主要表型中,咳嗽、慢性鼻炎、支气管扩张占93.9%,鼻窦炎占90.9%,中耳炎占45.5%,听力下降占21.2%,内脏转位占69.7%,不育明确记录的包括3例成年男性。回顾的记录详细的病例中,纤毛形态均发现异常。结论Minigene剪接变异体分析确定了DNAH5基因内含子区突变可影响该基因的剪接,提供了该突变位点致病性证据(PVS1)。DNAH5基因c.1731-18A>G和c.1933C>T(p.Gln645*)复合杂合变异可能是导致患者临床表型的原因。
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是一种罕见的纤毛运动障碍疾病,文献报道50多个基因变异可导致PCD的发生。PCD患者临床表型的非特异性使得PCD鉴别诊断困难[1, 2, 3, 4]。基因检测正成为一种新的诊断工具,大量的潜在致病变异基因被发现,大约1/3的致病变异基因会导致mRNA前体(pre-mRNA)剪接异常。人工合成包含基因特定区域的小型基因片段(Minigene)剪接技术可帮助确定非经典剪接位点的致病性,对患者的病因确诊提供关键依据[5]。本研究报道一家系中两例疑似PCD患者的临床和遗传学特征,家系全外显子测序和Minigene剪接变异体分析最终确诊了患者的遗传学病因,扩展了动力蛋白轴突重链5(dynein axonemal heavy chain 5,DNAH5)的基因变异谱。同时本文总结了中国人群中DNAH5变异导致PCD患者的常见临床表型。
对象与方法
1. 研究对象:患者家系图见图1。其中先证者(Ⅱ-2)为12岁男性,阵发性咳嗽、有痰不易咳出5年余,一直以“支气管肺炎”治疗。胸部CT示双肺多发微小结节影,较大者位于右肺下叶背段,约4 mm×3 mm,右肺中叶及双肺可见少许薄斑片状高密度影(图2)。心脏多普勒超声及肝胆胰脾肾超声检查未见异常。询问病史发现先证者姐姐(Ⅱ-1),27岁,自述间断咳嗽、咳痰、胸闷10余年,剖宫产生育一正常男婴。既往行鼻息肉切除术,因双肺支气管扩张行胸腔镜下肺叶切除术。心脏多普勒超声及肝胆胰脾肾超声检查未见异常。
 图1 患者家系图
图1 患者家系图
图2 先证者(Ⅱ-2)胸部CT 先证者2022年6月的CT结果显示双肺多发微小结节影,右肺中叶及双肺可见少许薄斑片状高密度影
2. 基因检测及致病性分析:采集全部家系成员外周血各3 ml,EDTA-K2抗凝。使用基因组DNA提取迷你试剂盒(德国Qiagen公司)提取外周血的总DNA。使用Qubit 3.0(美国Invitrogen公司)对样品DNA进行定量测定,质控合格后基因组DNA按照NanoWES技术平台标准流程获得全部外显子文库。经 Illumina Novaseq 6000二代测序平台对外显子文库进行PE150测序分析。测序数据过滤以等位基因频率<1%进行,人类基因组参考序列版本选择GRCh37/hg19。利用Primer5.0软件对可疑突变位点进行特异引物设计,Sanger测序(上海生工生物工程有限公司)验证患者的可疑致病位点。引物见表1。参照美国医学遗传学与基因组学学会变异指南对变异位点进行致病性评级。 3. Minigene剪接变异体分析:将DNAH5野生型和c.1731-18A>G(13内含子)突变体Minigene构建到pcDNA3.1中。2个重组载体分别转入HeLa和293T细胞系,转染后提取RNA。基因克隆及相关PCR引物合成由中国武汉百翼生物科技有限公司完成,观察该内含子变异是否影响外显子13、14和15的剪接。引物见表1。
3. Minigene剪接变异体分析:将DNAH5野生型和c.1731-18A>G(13内含子)突变体Minigene构建到pcDNA3.1中。2个重组载体分别转入HeLa和293T细胞系,转染后提取RNA。基因克隆及相关PCR引物合成由中国武汉百翼生物科技有限公司完成,观察该内含子变异是否影响外显子13、14和15的剪接。引物见表1。结果
1. 遗传学分析:全外显子测序检测到两例患者DNAH5基因(NM_001369)第72外显子存在c.12367C>T(p.His4123Tyr)变异(chr5∶13719123)、第13内含子存在c.1731-18A>G变异以及第14外显子存在c.1933C>T(p.Gln645*)变异。Sanger 测序证实两例患者的DNAH5基因c.12367C>T(p.His4123Tyr)和c.1731-18A>G变异遗传自父亲,而DNAH5基因c.1933C>T(p.Gln645*)变异遗传自母亲。见图3。
图3 家系成员DNAH5基因突变位点Sanger测序图,先证者之父(Ⅰ-1)携带c.12367C>T和c.1731-18A>G变异,先证者之母(Ⅰ-2)携带DNAH5基因c.1933C>T变异,先证者(Ⅱ-2)和先证者姐姐(Ⅱ-1)同时携带c.12367C>T、c.1731-18A>G以及c.1933C>T变异
2. Minigene体外试验结果:DNAH5基因c.1731-18A>G突变导致mRNA异常剪接,提前形成终止密码子。3种异常剪接情况:(1)DNAH5基因第13号内含子右侧滞留24bp,滞留的序列为CTTTTAGCTTTTTAAAAAAATCAG。最终在cDNA及蛋白水平的表示方式为DNAH5基因c.1730_1731insCTTTTAGCTTTTTAAAAAAAT CAG(p.Arg577Ser fs*3)变异,即突变后读码框改变,提前形成终止密码子。(2)DNAH5基因第14号外显子左侧缺失85bp。在cDNA及蛋白水平的表示方式为:DNAH5基因c.1731_1815del(p.Arg577Ser fs*47),突变后读码框改变,在第14号外显子内部提前形成终止密码子。(3)DNAH5基因第14号外显子跳跃。在cDNA及蛋白水平的表示方式为:DNAH5基因c.1731_2052del(p.Asn579Lys fs*5),突变后读码框改变,第15号外显子内部提前产生终止密码子。Minigene体外试验结果及序列保守性分析见图4。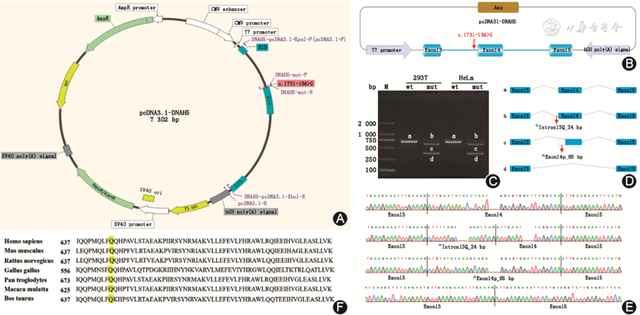
图4 患者DNAH5基因c.1731-18A>G突变的minigene体外试验检测结果及保守性分析 A和B:DNAH5-pcDNA3.1-野生型(wt)/突变型(mut)的质粒构建示意图;C:pcDNA3.1-野生型(wt)/突变型(mut)RT-PCR琼脂糖凝胶电泳图,在293T和HeLa细胞中,野生型只有一条a条带,突变型产生b、c、d三条带,且b条带较浅;D:minigene剪切示意图;E:剪切条带对应测序结果图,a为正常剪接条带测序图,b、c、d为3种异常剪接方式对应的3种测序结果图;F:DNAH5 p.Gln645在不同物种中序列保守图
3. 致病性分析:检出的DNAH5基因c.12367C>T(p.His4123Tyr)变异,gnomAD数据库中显示该突变位点在东亚人群中携带频率为0.01,ExAC数据库中该突变位点在东亚人群中携带频率为0.009。ClinVar数据库收录4例相同位点的变异。其中2024年1月29日最新评估3例该突变为良性或可能良性变异,而2018年1月12日评估1例该突变为致病性不明确。Mutation Taster软件预测该突变为良性。综合分析我们认为该突变为可能良性变异。
DNAH5 c.1933C>T突变,导致DNAH5第645位谷氨酰胺密码子突变成终止密码子,多肽链合成提前终止,对基因功能造成影响(PVSl)。该位点突变未在ClinVar、gnomAD及OMIM数据库中收录,且该序列在不同物种中序列保守(PM2_Supporting)。Minigene试验表明DNAH5基因c.1731-18A>G突变可能导致mRNA三种异常剪接,均造成读码框改变,提前形成终止密码子,导致多肽链合成提前终止,对基因功能造成影响(PVSl)。该突变未在ClinVar、gnomAD、千人基因组数据库及OMIM数据库中收录,且在中国知网、万方数据库、PubMed及Web of science等文献数据库中未见报道(PM2_Supporting)。家系验证表明两例患者的变异遗传自父母,为复合杂合变异。结合患者出现的临床表型如咳嗽、反复肺部感染,支气管扩张等(PP4)。根据美国医学遗传学与基因组学学会变异指南,两例患者携带的DNAH5基因c.1731-18A>G(PVSl+PM2_Supporting+PM3_Supporting+PP4)和c.1933C>T(PVSl+PM2_Supporting+PP4)突变被判定为致病性变异,与PCD相关,为常染色体隐性遗传。
4. 文献中已报道的中国人群中DNAH5变异导致的PCD患者表型总结:在万方医学数据库、中国知网和PubMed中以DNAH5为关键词检索,去除与主题无关的文献、外国人的报道及重复的病例,最终表型和基因型内容比较完整的因DNAH5突变导致PCD的中国人33例病例纳入整理。男性22例,女性10例,1例性别未知。咳嗽、慢性鼻炎、支气管扩张占93.9%(31/33),鼻窦炎占90.9%(30/33),中耳炎占45.5%(15/33),听力下降占21.2%(7/33),内脏转位占69.7%(23/33);病例中大多患者年龄不足18岁,在不孕不育的统计中,4例男性患者年龄超过20岁,其中3例明确不育(3/4)。在纤毛形态记录详细的22例病例中,1例纤毛动力蛋白臂正常但是纤毛排列紊乱,其他纤毛动力蛋白臂均存在缺陷或运动异常。具体见表2。

讨论
目前PCD的临床诊断主要依据患者临床症状,结合透射电子显微镜观察纤毛的超微结构结果或者基因检测PCD相关的致病基因突变。然而PCD本身临床表型缺乏特异性,部分PCD患者纤毛形态正常,且纤毛检测对机构医疗条件要求高,结果存在假阴性和假阳性[6, 7]。随着与PCD相关基因的不断发现,分子遗传检测正成为一种标准化的诊断手段,提高了疾病的检出率,利于疾病的鉴别诊断。全基因组测序技术可以在基因的编码区和非编码区发现大量的变异。临床上对于非经典区域的内含子突变,由于其致病性难以判断,容易被忽略。越来越多的文献报道剪接位点突变及内含子突变可导致异常的RNA剪接产物,可能对基因功能造成影响进而导致疾病发生。家系中两例患者表现出反复咳嗽、慢性鼻炎、支气管扩张等符合PCD的临床表型,全外显子测序结果显示两例患者均携带DNAH5的复合杂合变异。其中DNAH5基因 c.1933C>T(p.Gln645*)变异,使得突变位点引入了终止密码子,导致翻译提前终止。DNAH5基因c.1731-18A>G变异,虽然不是经典内含子变异,但是结合临床表型我们应该重视,需进一步分析其可能的致病性。Minigene技术可帮助分析隐蔽剪接位点的形成以及其对mRNA表达的影响,有效确定内含子突变位点的致病性。这里Minigene体外试验证实了该DNAH5内含子区突变可能导致3种异常剪接,均造成蛋白翻译提前终止。以上研究为确定DNAH5基因内含子区新变异的致病性提供了直接实验依据。根据美国医学遗传学与基因组学学会变异指南和标准,我们明确了DNAH5基因c.1731-18A>G和c.1933C>T(p.Gln645*)复合杂合变异可能是导致患者临床表型的原因。这两个新发现的基因突变位点,丰富了DNAH5基因的突变谱。遗传学检测技术的使用,有助于PCD的临床鉴别诊断。
2000年有学者首次报道位于5号染色体p15.2区域的DNAH5基因突变可导致PCD的发生[8]。目前已报道超过50个基因变异,包括DNAH11、DNAH5、DNAH1、CCDC39、CCNO、DNAI1、HEATR2,RSPH9等,与PCD相关[9, 10, 11]。不同基因突变导致的患者表型存在差异,不同种族之间表型差异也较大。DNAH5突变是导致PCD的最常见原因。这里我们回顾了2014—2024年发表文献中报道的中国人中DNAH5突变导致的33例PCD患者的表型与基因型[4,11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25]。结果显示反复咳嗽、支气管扩张和鼻窦炎的比例高达90%,与报道PCD患者的表型一致。临床诊疗中,对于反复咳嗽、咳喘的患者进行CT检测比较直观和清晰,同时还可检测是否存在内脏反位。我们统计的DNAH5突变导致PCD的患者中,内脏反位的比例也达到了69.7%。文献报道在内脏异位综合征患者及产前异位症的胎儿中均检测到DNAH5突变,这为DNAH5在内脏异位中的作用提供更多支持[26, 27]。对于中耳炎和听力下降表征,我们统计超过50%的患者未出现该症状,本文报道的两例患者也未出现此表型。体现了PCD患者的表型不特异性。病例中育龄期患者较少,虽然报道4例超过20岁的男性患者中,3例明确不育,但是DNAH5与不孕不育的相关性需进一步的研究。国内缺乏PCD长期随访的肺功能及影像学数据,PCD的进展速度也不尽相同,存在表现度差异,因此PCD的发病率可能被严重低估。在以后的诊疗工作中,对于有反复呼吸道感染、慢性鼻窦炎、支气管扩张的患者我们应高度警惕PCD的可能,同时要尽可能全面地收集患者异常表型并进行相关基因检测。研究显示PCD患者支气管扩张的比例随着年龄的增长而上升,肺损伤也会随着年龄的增长而增加。而目前PCD无特效疗法,主要以加强气道管理和抗感染为主。因此,早期诊断、及早进行健康指导和肺功能的康复锻炼对提高患者生活质量具有重要帮助。
参考文献(略)