
引言
细胞衰老这一概念最初由Leonard Hayflick及其同事于1961年提出,其在胚胎发育、癌症、衰老和其他多种疾病等生理病理过程中发挥着重要作用。细胞衰老具有多种特征,包括细胞周期阻滞、细胞周期蛋白依赖性激酶(CDK)抑制蛋白(如 p16Ink4a、p21CIP1)的表达、衰老相关-β-半乳糖苷酶(SA-β-Gal)活性升高,以及衰老相关分泌表型(SASP,包括一系列趋化因子、白细胞介素、生长因子或组织重塑蛋白酶等)的显现。精准靶向体内的衰老细胞对于揭示其在衰老和各种病理状况中的机理作用至关重要。目前,p16Ink4a是研究细胞衰老广泛使用的标志物之一,它在抑制CDK4/6活性和限制细胞周期从G1期向S期转变过程中发挥着关键作用。众多研究利用p16Ink4a启动子的激活来识别体内的衰老细胞,并构建了多种基于p16Ink4a的遗传小鼠模型。研究发现,在年轻和健康组织中基本上检测不到p16Ink4a的表达,但在衰老或损伤组织中会积累 p16Ink4a+细胞,通过遗传或药物方法消除p16Ink4a+细胞,可以延长早衰小鼠的寿命,减轻生理衰老小鼠的年龄相关表型,并改善多种疾病,如动脉粥样硬化、肺纤维化、糖尿病、肝脂肪变性以及肿瘤发生等。然而,也有研究表明,衰老细胞在某些情况下可能发挥积极作用,如肿瘤抑制、胚胎发育、伤口愈合、毛发生长、促进肺再生等。然而,目前的衰老细胞功能研究都是非特异性地针对所有衰老细胞,而不考虑细胞类型,这使得特定细胞类型衰老细胞的命运轨迹和生理病理作用尚不清楚。
2024年10月4日,中国科学院分子细胞科学卓越创新中心周斌研究员在Cell学术期刊在线发表题为Identifying specific functional roles for senescence across cell types的论文。该研究基于双同源重组酶系统建立了体内细胞衰老的谱系示踪及功能研究技术,系统探讨了肝脏损伤和修复过程中不同细胞类型衰老细胞的命运轨迹及其特定作用。这一研究工作不仅为肝脏疾病的临床治疗提供新的研究方向和理论依据,也为衰老领域和再生医学研究提供了新的技术路径与方法。

为了示踪体内衰老细胞及研究它们功能,研究人员首先构建了一种荧光报告基因小鼠品系p16-tdT,通过检测不同年龄段各器官中tdT的表达,发现tdT+细胞在不同器官中的类型多样且比例随着年龄的增长显著增加。同时,这些tdT+细胞表现出明显的SA-β-Gal活性、更低的增殖能力及高表达SASP因子等,进一步验证了p16Ink4a+细胞的细胞衰老表型。针对肝纤维化这一特定病理过程,研究人员利用四氯化碳(CCl4)诱导的肝脏损伤模型,结合免疫染色与scRNA测序技术,发现肝损伤中p16Ink4a+细胞主要涉及内皮细胞及巨噬细胞。通过基因表达谱分析,研究人员发现这些p16Ink4a+巨噬细胞与内皮细胞表现出独特的基因表达变化,包括SASP因子的表达和细胞衰老相关信号通路上调等。为了进一步研究细胞类型特异性p16Ink4a+细胞在肝损伤和修复中的命运,研究人员开发了一种名为Sn-pTracer(senescent cells-pulse-chase-tracer)的遗传系统,该系统利用了双重组酶技术,只有在共同表达Dre和Cre重组酶的细胞中,R26-RL-tdT(Rosa26-rox-Stop-rox-loxP-Stop-loxP-tdT)报告小鼠的两个Stop序列才会被移除,从而诱导tdT的表达。研究人员将巨噬细胞特异性F4/80-Dre或内皮细胞特异性Cdh5-DreER小鼠品系与p16-CreER和R26-RL-tdT小鼠交配,产生了F4/80-Dre;p16-CreER;R26-RL-tdT小鼠命名为SnMø-pTracer,和Cdh5-DreER;p16-CreER;R26-RL-tdT小鼠命名为SnEC-pTracer。在CCl4诱导的肝损伤中,SnMø-pTracer和SnEC-pTracer系统分别精确标记了p16Ink4a+巨噬细胞与p16Ink4a+内皮细胞。研究发现,损伤肝脏的p16Ink4a+巨噬细胞显著增多但修复后大量凋亡,被p16Ink4a–巨噬细胞替代。相反,p16Ink4a+内皮细胞在损伤后虽也增加,但在肝脏修复阶段大部分存活,表明其衰老状态对内皮细胞而言可能是可逆的细胞周期停滞。这些发现揭示了不同类型细胞在应对肝损伤时不同的衰老与修复机制,为深入理解细胞命运调控及组织修复策略提供了新视角。然而,使用Sn-pTracer系统需要研究人员具有细胞类型特异性的Dre工具小鼠,相对Cre工具小鼠数量还是比较少。此外,考虑到tamoxifen(Tam)在体内的血清半衰期较短,在损伤期间对Sn-pTracer小鼠持续使用Tam诱导以记录细胞衰老不仅是一种负担,而且可能对小鼠健康有害。为了克服这些限制,研究人员开发了一种新的遗传系统,命名为Sn-cTracer(senescent cells-Cre-induced-tracer),通过细胞类型特异性Cre启动持续记录p16Ink4a+细胞,从而使其更广泛地应用于体内p16Ink4a+细胞特异性靶向。根据设计,Sn-cTracer 涉及三种小鼠品系:细胞类型特异性Cre(ER)工具小鼠、p16-LSL-Dre(p16-loxP-Stop-loxP-Dre)和R26-RL-tdT-DTR(Rosa26-rox-Stop-roxP-Stop-loxP-tdT-DTR)。Cre-loxP重组可切除p16-LSL-Dre的Stop序列,从而在Cre表达细胞中将p16-LSL-Dre转化为p16-Dre,并且只有同时表达Dre和Cre的细胞才能移除R26-RL-tdT-DTR等位基因中的两个Stop序列,从而永久表达tdT和DTR(白喉毒素受体),实现以细胞类型特异性的方式对p16Ink4a+细胞进行不间断记录和遗传清除。在实验中,研究人员构建了SnMø-cTracer小鼠,用于探究p16Ink4a+巨噬细胞在肝纤维化中的作用。CCl4诱导肝纤维化损伤过程中,利用白喉毒素(DT)特异性清除这些细胞,发现其显著减轻了纤维化程度,揭示了p16Ink4a+巨噬细胞在纤维化过程中的促进作用。为进一步理解不同类型细胞在纤维化中的作用,研究人员还构建了SnEC-cTracer小鼠,聚焦于p16Ink4a+内皮细胞。与p16Ink4a+巨噬细胞相反,特异性清除p16Ink4a+内皮细胞后肝脏纤维化程度加剧,表明这些细胞在限制肝损伤和纤维化过程中发挥重要作用。Sn-cTracer系统不仅简化了实验流程,还提供了强有力的遗传工具来精准操控和解析p16Ink4a+细胞在复杂生理病理过程中的具体作用。为了深入理解p16Ink4a+巨噬细胞和内皮细胞在肝纤维化中的不同作用机制,研究人员采用scRNA-seq技术对SnMø-cTracer和SnEC-cTracer小鼠的肝脏非实质细胞进行了深入分析。在SnMø-cTracer小鼠中,DT处理显著改变了巨噬细胞群体的基因表达谱,上调了与免疫激活相关的通路,如促进自然杀伤细胞(NK细胞)和T淋巴细胞增殖,这提示清除p16Ink4a+巨噬细胞限制肝纤维化可能是通过改变了肝脏免疫微环境导致的。相比之下,SnEC-cTracer小鼠中DT处理导致间质细胞比例上升,表明肝纤维化加剧。内皮细胞群体的基因表达变化揭示了清除p16Ink4a+内皮细胞后,肝组织向促纤维化环境转变,成纤维细胞和间充质细胞增殖增强,提示p16Ink4a+内皮细胞抑制成纤维细胞增殖的重要作用。这些发现不仅揭示了两种细胞类型在肝纤维化中的不同作用机制,也为开发针对肝纤维化的精准治疗策略提供了新的见解。通过RNA-Seq分析,研究人员发现在血管生成与肝脏修复中发挥重要作用的VEGF信号转导受体Kdr表达在p16Ink4a+内皮细胞中有下调趋势。鉴于VEGF信号对内皮功能及肝脏健康的关键作用,研究人员开发了Sn-gTracer系统(senescent cells-gene manipulation-tracer),旨在细胞类型特异性地调控p16Ink4a+细胞中基因表达。根据设计,Tam诱导的Cdh5-DreER(用于确定细胞类型,在本例中为内皮细胞)将内皮细胞中p16-CreXER(p16-Cre-rox-ER-rox)转换为p16-Cre,随后实现靶向任何Cre/loxP系统介导的等位基因敲除或者过表达,同时允许表达Cre介导的报告基因来示踪细胞类型特异性衰老细胞。研究人员利用SnEC-gTracer系统实现了在p16Ink4a+内皮细胞中Kdr的过表达。在肝纤维化模型中,SnEC-gTracer小鼠肝脏内具有较多mCherry+内皮细胞,并伴随EdU结合阳性,表明具有一定的细胞增殖能力,同时SASP因子表达减弱。更重要的是,纤维化标记物免疫荧光染色及天狼星红染色结果均显示,Kdr过表达显著减轻了肝纤维化程度。这一发现不仅揭示了p16Ink4a+内皮细胞在肝纤维化中的新角色,还展示了通过遗传重编程改善肝脏损伤的巨大潜力,为肝纤维化治疗提供了新的策略与希望。综上,细胞类型特异性p16Ink4a+细胞的精准遗传靶向是揭示其在衰老与疾病中功能的关键,研究人员建立了体内细胞衰老的谱系示踪及功能研究技术,开发了四种互补的遗传策略,研究不同细胞类型中p16Ink4a+衰老细胞的体内命运与特定功能,不仅为细胞衰老与损伤再生的研究提供了新的视角和工具,也为未来精准靶向细胞衰老疗法奠定了坚实的理论基础。
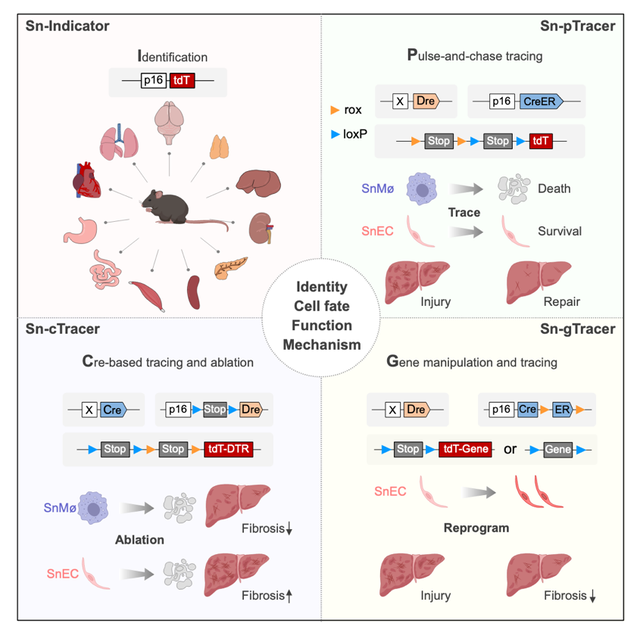
利用体内细胞衰老的谱系示踪及功能研究技术揭示不同细胞类型衰老细胞特定功能(Credit: Cell)
参考文献
https://doi.org/10.1016/j.cell.2024.09.021
责编|探索君
排版|探索君
文章来源|“BioArt”
End
