麝香由成年雄鹿分泌,是一种极具价值的中药和天然调味剂,因其高的医疗和经济价值而倍受珍视。
长期的非法狩猎和栖息地的碎片化导致中国野生麝香的数量在过去70年里迅速下降了97%以上,从20世纪50年代估计的200-300万头下降到2009年的约73,480头。
中国拥有世界上最大、分布最广的麝香种群,占全球麝香总数的70%以上,同时也贡献了世界麝香总产量的90%以上。肠道微生物区系对宿主的健康、生存和环境适应是必不可少的。
然而,摄食环境和食物的变化会影响麝香肠道微生物区系的组成和功能,最终影响它们的健康和适应。
保持健康的状态对濒危高山麝香(Moschus chrysogaster,AMD)圈养繁殖的成功至关重要,圈养繁殖有利于该物种的迁地保护和野生种群恢复。

在中国所记录的6种麝中,高山麝体型最大,野生AMD的种群估计约为28,000只,被列为世界自然保护联盟红色名单上的濒危(EN)和中国红色名单上的极度濒危(CR)。
主要分布在青藏高原和中国周边地区,包括西藏东南部、青海、祁连山脉和甘肃兴隆山、宁夏贺兰山、四川西部和云南北部的高海拔草甸、灌丛或针叶林。
为了获得珍贵的麝香,中国于1958年开始了人工饲养麝香的计划,作为一种替代捕杀麝香的选择,这被认为是迁地保护野生麝香资源和可持续利用麝香的有效措施。
在过去的60年里,人工饲养麋鹿的规模逐渐扩大,特别是AMD,使其成为中国第二大人工繁殖麋鹿项目。
甘肃兴隆山国家级自然保护区于20世纪90年代开始驯养麋鹿,目前拥有中国最大的麝场。
肠道微生物区系是宿主肠道微生态的重要组成部分,在维持肠道内环境稳定、促进宿主健康、调节新陈代谢、营养吸收、生长发育等生理过程以及免疫调节和抵抗外来病原体方面发挥着至关重要的作用。
与野生麝鹿相比,圈养个体对肠道疾病的易感性更高,包括胃肠炎、腹泻和胃肠道出血,这些疾病往往与细菌调节失调有关,从而导致更高的死亡率,并构成人工饲养规模扩大的主要限制因素
目前,人工饲养驯养大马鹿的成功为野生放流和可持续利用麝香资源奠定了基础。
圈养动物肠道微生物区系是宿主肠道微生态的重要组成部分,在维持肠道内环境稳定、促进宿主健康、调节新陈代谢、营养吸收、生长发育等生理过程以及免疫调节和抵抗外来病原体方面发挥着至关重要的作用。
利用16S rRNA基因测序方法,从不同的分类水平研究了圈养和野生麝肠道微生物区系的组成和多样性。
重点解决:(I)确定麝香的核心微生物区系;(Ii)确定圈养和野生麝间肠道微生物组成的差异;(Iii)分析优势菌和代谢功能的差异;以及(Iv)调查圈养和圈养麝间条件致病菌和疾病相关功能的变化。
本研究的结果将通过分析圈养种群和野生种群肠道微生物区系的差异,为制定有效的麝类健康管理策略提供科学依据。
 样本获取
样本获取在青海省互助县北山国家森林公园,用非侵入性采样方法从野生AMD中采集了23份新鲜粪便样本(图1)。
此外,2021年3月10日和11日,在甘肃省榆中县兴隆山国家级自然保护区的AMD繁育中心,从圈养个体身上采集了25份粪便样本。
在对圈养的个体进行采样之前,AMD繁殖场进行了彻底的清洁,每个个体都被限制在一个个体的养殖场,以便第二天早上能够收集新鲜的粪便。
在采样过程中,在排便后立即使用无菌一次性聚乙烯手套和无菌采样袋采集新鲜粪便样本。
图1 野生(红三角)和圈养(黑三角)高山香鹿采样点(AMD)
OTU聚类和分类注释从体重200毫克的麝香中无菌采集新鲜粪便,并将其转移到2毫升无菌离心管中。
Omega Bio-Tek的土壤DNA试剂盒根据制造商的说明从野生和圈养的AMD粪便样本中提取基因组DNA。
用1%琼脂糖凝胶电泳法评价DNA提取的质量,用NanoDrop2000测定DNA的纯度。
用2%琼脂糖凝胶电泳法对纯化的扩增产物进行分析,并用Quantus™荧光仪进行定量。
使用NEXTflex®Rapid DNA-Seq Kit为测序准备文库。
将扩增出的片段串联连接,磁珠筛选去除自连接序列片段。得到的文库被纯化并回收用于在Illumina MiSeq PE300平台上测序。原始测序数据保存在NCBI SRA数据库。
为了处理原始测序数据,使用Trimomatic软件(v.0.39)筛选末端质量评分低于20的低质量碱基,并使用Flash软件(v1.2.7)合并成对的末端读数。
使用UPARSE软件(v7.1)对所有具有97%相似性的样本进行OTU聚类,并且在聚类过程中去除嵌合序列。
使用核糖体数据库项目(RDP)分类器(http://RDP)对每个有效序列的物种分类进行注释。
生物信息学分析在对OTUS进行分类标注后,统计每个样本中每个OTU标注对应的丰度信息,并根据样本序列的最小数目对样本序列进行二次采样。用R软件用群落直方图和维恩图显示了野生和圈养AMD在门和属水平上的所有样本或每组样本的丰度。分析了微生物组成的相似性和差异性,并绘制了丰度图。利用R软件绘制了野生(红三角)和圈养(黑三角)高山香鹿(AMD)应用微生物学和生物技术1-3直方图和热图。
肠道微生物群落组成经质量控制后,48份粪便样品共获得528,7059个原始读数,平均每个样本获得110,147个读数,平均读数长度为375个碱基对。
经过97%的相似性聚类后,共鉴定出2105个有效的OTUS,其中野生和圈养AMD共享1716个OTUS,野生和圈养个体分别拥有152个和237个OTUS。
经鉴定的OTUS隶属于12门21纲52目97科212属。
Good‘s盖度指数表明,野生和人工饲养的麝肠道微生物区系都具有很高的代表性,盖度指数都在99%以上。
根据物种注释,发现野生AMD和圈养AMD的相对丰度均大于1%的优势类群为菲米鼠(野生,64.2%;圈养,82.1%)和拟杆菌(野生,33.9%,圈养,16.3%)。
在可识别的前50个细菌属中,UCG-005、Christensenellaceae R-7群、拟杆菌属、Monoglobus、反胃球菌、Prevoteaceae UCG-004和Rikenellaceae RC9肠群是可识别的优势属,野生和圈养AMD的相对丰度大于1%(图2B)。
此外,在野生个体中发现的其他优势属包括玫瑰草属,而在圈养个体中发现的优势属包括NK4A214组属和泽泻属,均属于拟杆菌门。
聚类结果表明,野生个体和圈养个体分别聚为一类。
野生与圈养AMD肠道微生物区系差异分析:野生AMD肠道微生物区系的α多样性显著高于圈养个体(图3A),这是由四个阿尔法多样性指数衡量的:SOBS、Shannon、Chao1和系统发育多样性(PD)(图3A)。
PCoA和ANOSIM还表明,野生和圈养AMD的肠道微生物组成存在显著差异,R值分别大于0.001和P值(图3B)。
这些结果表明,野生和圈养AMD之间的肠道微生物区系组成的组间差异显著大于组内差异。

图2.野生和圈养AMD肠道微生物群落组成。
图2中(A)为细菌门相对丰度的直方图分析,(B)是基于前50个属细菌的热图分析。
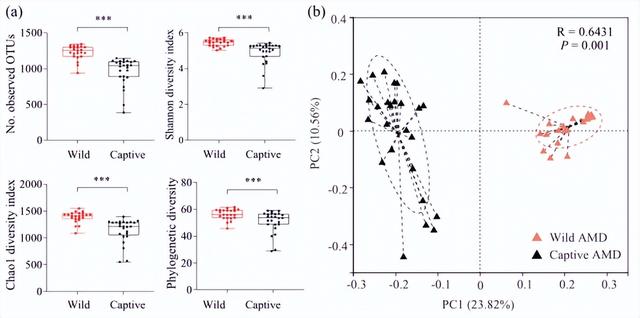
图3.野生和圈养AMD肠道微生物区系多样性和组成的比较。
图3中,(A)基于SOBS、Shannon、Chao1和系统发育多样性(PD)指数的α多样性分析。(B)基于Bray-Curtis距离矩阵的主坐标分析(PCoA),显示WARD和WARD之间的差异。
肠道微生物区系优势菌及代谢功能的差异野生AMD与圈养AMD相比,野生AMD和圈养AMD的拟杆菌和野生AMD的相对丰度均有显著差异。
野生个体比圈养个体中的菲米库斯数量明显更多,而拟杆菌门则表现出相反的模式(图4A)。此外,UCG-005属的丰度、千金藤科R7群。
与圈养个体相比,野生AMD患者的单胞菌、瘤胃球菌和玫瑰香杆菌数量显著增加(P<0.05)(图4B)。
相反,圈养个体类杆菌属、Rikenellaceae RC9肠组、NK4A214组和泽泻属的丰度显著高于野生个体(P<0.05)。在野生和圈养个体之间,普氏原虫属UCG-004的丰度没有任何显著差异。
根据KEGG数据库的功能注释分析,野生和圈养AMD的肠道微生物区系主要执行代谢功能。
重点是碳水化合物代谢(X̅=10.31%)、氨基酸代谢(X̅=9.85%)、能量代谢(X̅=5.84%)、辅因子和维生素代谢(X̅=4.34%)、核苷酸代谢(X̅=4.22%)、脂肪代谢(X̅=2.87%)、糖类生物合成和代谢(X̅=2.01%)、萜类和多酮代谢(X̅=1.65%)。
异源生物的生物降解和代谢(X̅=1.62%)、其他氨基酸的代谢(X̅=1.41%)和其他次生代谢产物的生物合成(X̅=0.93%)。
KEGG数据库水平1和水平2的功能差异分析表明,野生AMD与各种营养和能量相关的代谢功能显著高于圈养个体(图4C,d)。

图4.野生与圈养AMD肠道微生物区系优势菌及代谢功能的差异分析。
在图4中,主要是优势菌门细菌(A)和优势属细菌(B)的差异分析,基于KEGG(京都基因和基因组百科全书)1级(C)和2级(D)数据库以及eggnog(基因进化谱系:非监督同源群体)数据库(E)的代谢功能差异分析。
致病菌及疾病相关功能的差异Wilcoxon秩和检验分析野生和圈养AMD之间潜在的条件致病菌的差异。
在门的水平上,圈养个体比野生个体显示出更高的放线杆菌和螺旋藻的相对丰度(图5A),而野生群体中变形杆菌的相对丰度更高。
在属水平上,圈养个体与野生个体相比,臭杆菌、链球菌、Oscillibacter、密螺旋体、感觉梭菌1、感觉梭菌6、副杆菌、泰泽氏菌、芽孢杆菌、气球菌和棒状杆菌的相对丰度较高(图5B)。
相比之下,白僵菌属、志贺氏菌属、厌氧菌属、消化球菌属、放线菌属和变色杆菌属在野生群中含量较高。
KEGG数据库中的功能注释分析表明,在水平1,圈养AMD肠道微生物区系的疾病相关功能丰富程度显著高于野生个体(图5C)。
在第2级,与传染病、代谢性疾病、癌症、神经退行性疾病和免疫系统疾病相关的疾病相关功能在圈养个体中显著高于野生个体(图5C)。

图5.在门一级(A)和属一级(B)对机会性病原体进行差异分析。(C)根据KEGG数据库对疾病相关功能进行差异分析。
核心细菌的共生网络共生网络分析揭示了野生和圈养AMD之间肠道微生物区系的复杂性和稳定性的差异。
野生组的环节较多,且负相关与正相关的比率高于圈养组,说明野生AMD的肠道微生物区系比圈养AMD更复杂和稳定(图6)。

图6.野生和圈养AMD中前50个细菌属的共生网络。
不同环境下高山麝肠道菌群影响尽管圈养繁殖被认为是保护濒危野生动物的最有效策略之一,但长期圈养可以显著改变生活环境和食物组成,导致肠道微生物区系的组成和功能发生变化。
因此,肠道微生物多样性的减少可能会增加潜在致病菌的比例,并丰富与疾病相关的功能,对圈养个体的福祉构成风险。
为了改善圈养麝鹿的健康状况,我们建议增加它们的天然食物摄入量,同时减少它们对工厂化饲料的依赖。
此外,我们建议模拟麝香在圈养环境中的自然生活条件,以帮助维持健康的肠道微生物区系。
肠道微生物区系与宿主共同进化,是宿主肠道微生态系统的重要组成部分,在调节宿主新陈代谢、生长、发育、免疫功能、病原体防御、生态适应和进化方面发挥关键作用。
年龄、遗传背景、性别、食物、季节、地理和饲养环境。分析野生和圈养AMD肠道微生物组成和功能的差异对于它们在野外的成功释放和种群扩张是至关重要的。
V4-V5可变区被认为是分析细菌16S rRNA基因的最佳区域,因为它表现出最小的基因组异质性,并有效地捕获基因组间变异。
圈养的拟杆菌门和拟杆菌属、Rikenellaceae RC9肠道群、NK4A214群和泽泻的相对丰度显著高于野生AMD。
众所周知,以上这些都是通过提高寄主的适应性而使寄主受益的。类杆菌是一种常见的有益细菌属,参与调节胆汁酸、短链脂肪酸、糖、蛋白质和脂肪的代谢。
参考文献【1】高山麝病株兔出血症病毒分离株的研究
【2】神经性厌食症和肠道微生物区系:汇集微生物数据的系统回顾和定量合成。
【3】基于高通量测序的黄芩提取物对热证大鼠肠道菌群多样性的影响。
【4】 神经性厌食症患者体重增加不能改善粪便微生物区系、支链脂肪酸分布和胃肠道症状。
