开发高效的电催化剂用于酸性电合成过氧化氢(H2O2)具有重要意义,而大多数材料在酸性条件下的选择性和稳定性会受到严峻挑战。
厦门大学黄小青教授、华中科技大学杨利明研究员等人证明了在结晶Pt核上构建无定形铂硒(Pt-Se)壳层可以操纵氧还原反应(ORR)途径,从而有效地催化酸性介质中H2O2的电合成。优化壳厚的Se2-Pt纳米粒子对H2O2的选择性超过95%,同时抑制了H2O2的分解。在流动反应器中,Se2-Pt纳米颗粒能在250 mA cm-2的电流密度下保持400 h,产生的H2O2浓度为113.2 g L-1,产率为4160.3 mmol gcat-1 h-1,有效降解有机染料。
所构建的无定形Pt-Se壳层获得了理想的O2吸附模式,提高了选择性,并诱导应变以优化OOH*结合,加速了反应动力学。这种硒化方法可推广到其他贵金属催化剂的设计,并提升2e- ORR性能。


针对原创性,作者进行了如下解释:在本文中,重点超越了材料合成,包括优化非晶结构的结构控制策略。作者引入了一种通用的硒化策略,该策略诱导贵金属纳米材料的应变和相变,增强了酸性环境下2电子O2还原成H2O2的能力。晶核与非晶壳相互作用产生的应变细化了非晶壳中孤立位点的电子结构,使其比低应变结构更有效地产生H2O2。
此外,从小规模的H2O2电合成扩展到模拟酸性电解质连续生产的实际条件。值得注意的是,最佳催化剂在流动反应器中以250 mA cm-2持续产生H2O2超过400小时。所得溶液具有较高的H2O2浓度,在实际应用中可以有效地去除有机染料。最后,作者证明了硒化方法适用于其他贵金属的2电子ORR通路。因此,相信这项工作为设计H2O2电合成材料及其他领域提供了新的视角,所有这些优势都值得在《Nature Communications》上发表。


作者采用两步后修饰法制备了厚度可控的结晶Pt核和非晶Pt-Se壳组成的核壳纳米颗粒(图1a)。具体而言,将Pt NPs作为生长晶种,引入Se可以在表面构建无定形Pt-Se壳。
值得注意的是,界面处的结构失配进一步诱发了应变相互作用(图1a)。可以通过改变硒的引入量(Sex-Pt NPs,x表示硒化程度)来精细调节非晶壳的厚度,从而实现对表面结构的连续调制(图1b)。
然后进行了一系列表征,以展示具有不同壳厚度的Se-Pt NPs的结构演变。从XRD图中可以清楚地观察到,随着硒化程度的加深,从Pt NPs到Se-Pt NPs在(111)峰(~ 39°)处的峰强度持续衰减,表明NPs的结晶度随着非晶Pt-Se壳层厚度的增加而继续降低(图1c)。相应的,SEM-EDX结果显示Se元素呈持续增加的趋势(图1d)。
为了明确核壳结构的表面种类,进行了仅探测最外层原子层的低能离子散射(LEIS)。从LEIS光谱中可以观察到,Se2-Pt NPs比Pt NPs显示出额外的Se信号,表明表面的非晶壳由Pt和Se元素组成(图1e)。利用XAFS证实了核-壳结构的结构演变。
如图1f所示,与纯Pt NPs相比,非晶壳层较厚的核壳催化剂的峰值强度不断降低。结合拟合结果,具有较厚非晶壳层的Se-Pt NPs的Pt-Pt键配位数减少,而Pt-Se键配位数增加,表明硒化程度加深。此外,小波变换计数图中峰位的负移表明,由于配位Se原子的原子序数较小,Pt-Se配位更多,这支持了Se-Pt NPs中Pt原子的配位环境演化(图1g)。

通过TEM进一步研究了具有可调壳厚的Se-Pt NPs的结构。首先,对应的映射图显示了Se元素向Pt元素核心的空间分布逐渐加深,证明了Pt-Se壳层厚度的演化过程(图2a-d)。从不同硒化阶段的TEM图像(图2e-h)可以看出,硒化过程是由外向内进行的,初始阶段以硒原子在Pt核表面掺杂开始(图2f)。
随着反应的进行,在Pt核周围形成一层薄薄的非晶态Pt-Se壳层,非晶态壳层的厚度逐渐变厚(图2g)。最后,可以得到由晶态核和非晶态壳组成的核-壳结构纳米材料(图2h)。 通过循环伏安(CV)技术对纳米材料的电化学行为进行研究,进一步揭示了纳米材料硒化的不同阶段。
如图2所示,Pt NPs的CV曲线在0.05~0.40 V之间呈现出典型的氢吸附/解吸峰,而Se-Pt NPs则呈现出一个不断减弱的氢吸附/解吸峰,且硒化程度更深。因此,可以合理地推断,随着硒化的进行,表面的Pt原子逐渐转变为孤立的位点,直到形成非晶壳层,进一步的硒化会导致Se原子插入更深的Pt晶格中,这与TEM结果一致(图2e-h)。 几何相位分析的应变映射显示,代表应变程度的颜色强度在整个Pt NPs中变化很小,表明结构内部的应变可以忽略不计(图2i)。
随着非晶壳的逐渐形成,应变产生并逐渐增加(图2j、k),并达到最大值,直到形成过厚的非晶壳(见图2l)。相应的,界面附近Pt晶格的强度分布图呈现逐渐的正位移(图2n),表明结晶Pt核存在拉伸应变。

通过RRDE测量,探索了不同壳厚Se-Pt NPs在O2饱和的酸性介质中的2e- ORR性能。如图3a所示,记录的LSV曲线显示了在0.1 M HClO4中在圆盘电极上测量到的O2还原电流(实线)和在Pt环电极上检测到的H2O2氧化电流(虚线)。因此,Se2-Pt NPs的H2O2选择性在很宽的电位范围内(0.0 V-0.6 V)>95%,表明一个高度活跃和选择性的2e-途径,转移电子数接近~2.0(图3b)。
相比之下,硒化程度较低的Se1-Pt NPs表现出相对优势的4e-通路(H2O2<36%,n~3.5),而较厚的NPs虽然表现出2e-通路的特征(H2O2>70%,n~2.6),但表现出明显较大的过电位和较低的环电流(图3a、b),揭示了2e- ORR性能与非晶壳厚度之间的相关性。
重要的是,Se2-Pt NPs还可以在各种酸性电解质中表现出对2e- ORR的高选择性,包括0.1 M H2SO4(≈95.6%)、0.5 M HClO4(≈96.1%)和0.5 M H2SO4(≈96.3%),在更宽的电位范围内优于大多数报道的催化剂(图3c)。
对于H2O2电合成的实际应用,有利的2e-途径和分解反应的抑制对于理想的催化剂是必不可少的(图3d)。系统地研究了所制备的催化剂在歧化反应和电化学还原反应中的活性。从电化学还原反应中H2O2还原电流明显降低(图3e)和歧化反应中H2O2分解受到抑制(图3f、 g)的结果可以看出,非晶壳的构建可以显著抑制H2O2的热分解和电化学分解反应。
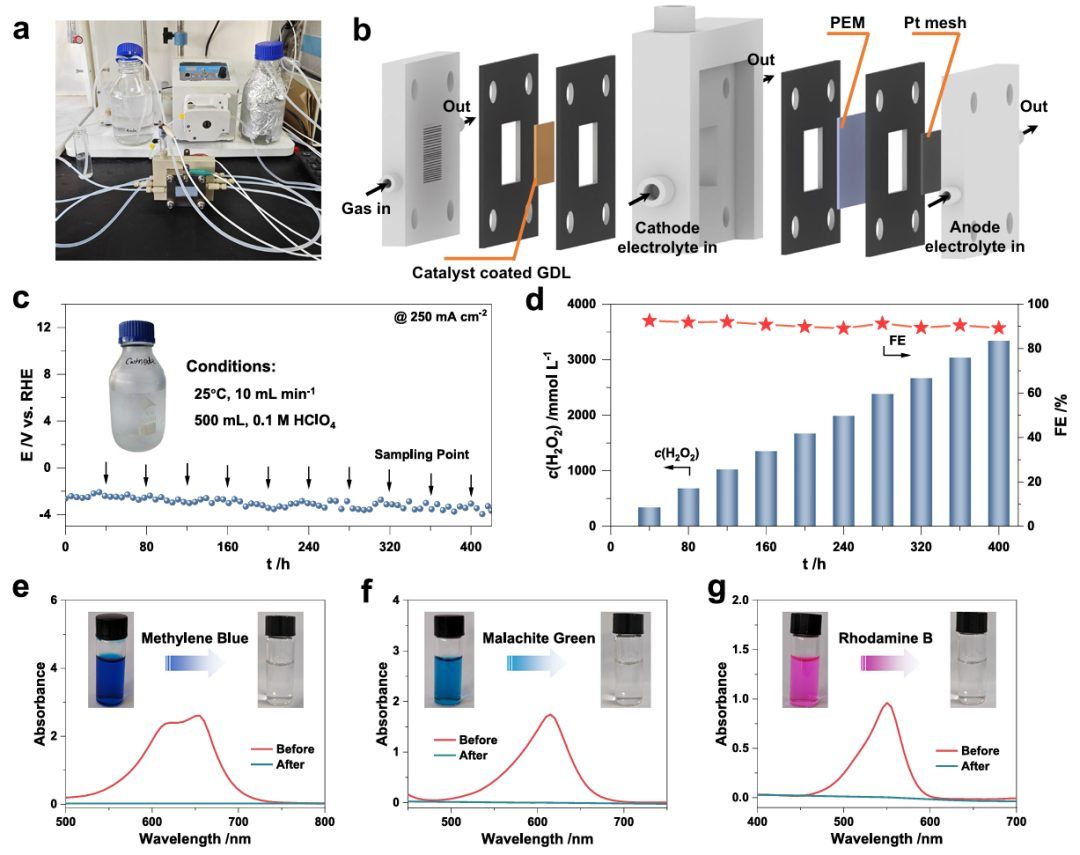
受Se2-Pt NPs良好的催化性能和抑制H2O2分解反应的启发,进一步将该催化剂引入实际的流动反应器中,在高电流密度和高体积下连续在酸中产生H2O2(图4a)。
图4b显示以Pt网为阳极,含Se2-Pt NPs的气体扩散层为阴极的流动反应器。在流动反应器中电合成H2O2的操作条件(如工作温度、液体流速、电解质环境和体积)如图4c插图所示。在连续O2流下,在250 mA cm-2的高电流密度下收集的极化曲线显示出稳定的状态,表明Se2-Pt NPs在运行条件下具有实际应用的潜力(图4c)。

作者进行DFT计算,进一步了解硒化程度对催化性能和选择性的影响。计算出的TDOS和PDOS如图5a、b所示,揭示了这四种催化剂的金属性质,它们具有优异的电子导电性,这是促进快速电荷转移和促进ORR的关键因素。
此外,催化剂和中间体之间的轨道重叠和杂化表明了有效的电荷转移和成键相互作用的形成。根据四种对比催化剂在选定电位下的ORR自由能图,可以推断,Pt NP和Se1-Pt NP都表现出4e-途径,因为由于存在连续的活性位点,*O*OH倾向于解离。相反,硒含量增加的Se2-Pt NPs和Se3-Pt NPs在其表面显示出孤立的Pt位点。
因此,O2以端对端方式吸附,O-O键不易断裂,从而形成H2O2。 根据图5c的计算结果,可以推断出Se2-Pt NPs和Se3-Pt NPs上2e-(4e-)途径的反应垒分别为0.42(0.91)eV和0.79(2.06)eV。两种催化剂中2e-途径的势垒均明显低于4e-途径的势垒,表明2e-途径是主要的反应过程。
此外,与Se3-Pt NPs相比,Se2-Pt NPs上2e-途径的势垒甚至更小,从而证明了Se2-Pt NPs在H2O2生成中的优越催化性能。 更重要的是,为了深入了解硒化对活性的应变效应,使用不同的表征工具,从不同的角度对催化剂、中间体和活性位点进行了深入的电子结构分析。非晶Pt-Se壳层与结晶Pt核界面处不同物体上的电荷密度分布如图5d-f所示。具体而言,当形成适当厚度的非晶壳层时,界面与活性位点Pt原子之间的电荷转移达到最佳状态,这对活化含氧中间体起着重要作用。
然而,随着Pt-Se非晶壳层厚度的增加,界面电荷转移的影响逐渐减弱。 然后,进一步计算了活性位点Pt原子的d带中心,以阐明各种界面对表面活性位点的影响(图5g)。结果表明,活性位点Pt原子的d带中心为εd(Pt)>εd(Se1-Pt)>εd(Se3-Pt)>εd(Se2-Pt),非晶壳层厚度对εd(Se2-Pt)有重要影响,与实验结果一致。
因此,随着硒含量的增加,形成无定形Pt-Se壳,并产生相应的应变效应。适当的壳层厚度可使非晶壳层与晶核之间的相互作用最强,从而优化电荷转移和表面Pt位点的d带中心。

如图6a所示,与Pd NPs相比,硒化Pd NPs表现出非晶壳形成的特征,表明成功构建了由晶核和非晶壳组成的结构(Se-Pd NPs)。有趣的是,与Pd NPs相比,Se-Pd NPs表现出主导的2e-还原途径,其环电流显著提高(图6b)。
此外,该策略是通用的,甚至可以扩展到商业催化剂(商业Pt/C和商业Pd/C)(图6a)。图6c表明,非晶壳的构建可以显著调节商用催化剂的还原途径。具体来说,Se-Pd NPs在0.15至0.47 V之间的H2O2浓度明显高于80%,而Pd NPs的H2O2浓度为~0%;Se-Pt/C和Se-Pd/C的H2O2浓度从~0%至~55%和~7%至~67%也有明显提高(图6d)。此外,根据统计结果(图6e)可以推断,核壳结构的构建使纳米材料从主要的4e-反应途径催化剂转变为以2e-反应途径为主的催化剂,显示了其对ORR途径的调控以及硒化诱导的应变和相变的普遍性。
文献信息
Selective and durable H2O2 electrosynthesis catalyst in acid by selenization induced straining and phasing,Nature Communications,2024. https://www.nature.com/articles/s41467-024-53607-5