
在工业上,乙炔(C2H2)半氢化为乙烯(C2H4)过程可以用于从烯烃中去除微量C2H2,也可用于更高要求的(E)-烯烃的合成。对于常用的炔烃半加氢催化剂,廉价的镍基和铜基催化剂表现出优异的催化性能。然而,其在低温下很难达到与Pd催化剂相同的高C2H4生成活性和选择性。此外,多相催化中的亚表面化学在调节催化性能方面起着重要作用。
为了阐明亚表面杂原子的作用,太原理工大学王宝俊、章日光等人通过密度泛函理论(DFT)计算和微动力学模拟,对一系列掺杂亚表面杂元素H、B、C、N、P或S的钯催化剂上的C2H2半加氢进行了全面研究。计算方法与模型作者使用Materials Studio 8.0中的Dmol3包进行DFT计算,并采用广义梯度近似(GGA-PBE)泛函来描述交换关联作用,以及采用有效芯电势(ECP)基组来处理金属Pd原子,而全电子基组用于处理非金属元素。
作者采用双数值极化(DNP)基组来扩展价电子函数,并采用DFT-D校正方法考虑范德华效应,以及对布里渊区使用3×3×1 Monkhorst–Pack k点采样。作者采用LST/QST方法搜索基本反应的过渡态,并通过振动分析和TS确认的计算来验证了过渡态的合理性。
由于(111)晶面是Pd催化剂的主要暴露和稳定的晶面,作者采用每层具有16个Pd原子的四层p(4×4)表面来模拟Pd催化剂,并且将杂原子H、B、C、N、P或S掺杂到Pd催化剂的亚表面中,以模拟亚表面掺有杂原子(H、B,C、N,P或S)的一系列Pd催化剂,其中顶部两层Pd原子和亚表面原子保持驰豫。在垂直方向上设置15Å真空层,以避免板层之间的周期性相互作用。
在此,本研究考虑了由亚表面杂原子掺杂的Pd催化剂,对应于1/16、1/2和1.0ML的覆盖率的催化剂,分别命名为Pd-A、Pd-A0.5和Pd-A1.0(A=H、B、C、N、P或S)催化剂。
结果与讨论

图1. 亚表面杂原子掺杂钯和钯催化剂的优化结构
如图1所示,在不同覆盖率下的亚表面H原子掺杂结构中,由于掺杂一个H原子和八个H原子对Pd的几何结构几乎没有影响,因此作者将16个H原子掺杂到亚表面中。其次,作者还考虑了1/16和1/2ML的B覆盖率下的掺杂。最后,对于较大半径的N、P或S原子,具有1/2ML亚表层N、P和S掺杂的优化结构具有明显的表面重构(图1);在1.0 ML的亚表面N、P或S覆盖下,1.0 ML N掺杂的优化结构已经变形。
同时,虽然亚表面P或S覆盖率为1.0ML的优化结构没有变形,但在吸附C2H2后结构明显变形,因此仅考虑了亚表面N、P或S原子1/16 ML覆盖率的掺杂结构。

图2. C2H2半氢化和聚合过程中可能涉及的途径
如图2所示,C2H2半氢化包括两个过程:氢化包括三条路线:C2H4脱附路线、C2H4加氢路线和CHCH3加氢路线。C2H4脱附路线有望实现高C2H4选择性,而由于乙烷的产生,其他两条路线需要被抑制。而对于聚合过程,首先是C2H2+C2H2生成C4H4,然后是连续氢化至C4H6,称为C2H2+C2H2耦合路线。第二种是C2H2+C2H3耦合路线,即C2H2+H→ C2H3,然后是C2H3+C2H2→C4H5,第三种是C2H3+C2H3耦合路线,即C2H2+H→ C2H3,然后是C2H3+C2H3→ C4H6。

图3. 393K下C2H2和C2H4在亚表面杂原子掺杂Pd催化剂上的吸附吉布斯能

图4.(a)C2H4氢化生成C2H5的活化吉布斯能和C2H4解吸吉布斯能。(b)393K下C2H4脱附路线和CHCH3加氢路线的总势垒
如图3所示,C2H2在11种类型的Pd-H、Pd-H0.5、Pd-H1.0、Pd-B、Pd-B0.5、Pd-C、Pd-C0.5、Pd-N、Pd-P、Pd-S和Pd催化剂上的吸附强度远大于C2H4。如图4a所示,在Pd(93.5 vs 95.4 kJ mol–1)和Pd-H0.5催化剂(90.1 vs 89.7 kJ mol-1)上,C2H4解吸与C2H4氢化在能量上具有竞争性。与在Pd-H(88.5 vs 99.0 kJ mol–1)、Pd-B(87.0 vs 100.2 kJ mol-1)和Pd-C催化剂(84.6 vs 97.2 kJ ol–1)上进一步氢化相比,C2H4脱附的动力学稍好。
因此,与Pd催化剂相比,Pd-H、Pd-H0.5、Pd-B和Pd-C催化剂不能显著促进C2H4脱附并抑制C2H4氢化为C2H5。因此,亚表面H、B和C 1/16 ML覆盖率掺杂和1/2 ML的亚表层H覆盖率掺杂的Pd仍然具有低的C2H4选择性。从动力学上讲,六种催化剂上的C2H4脱附比氢化更容易发生,其中包括Pd-H1.0(80.7 vs 122.5 kJ mol–1)、Pd-B0.5(57.2 vs 144.6 kJ mol-1)、Pd-C0.5(73.0 vs 166.3 kJ mol-1)、Pd-N(95.0 vs 147.0 kJ mol–1),Pd-P(81.0 vs 105.4 kJ mol-1)和Pd-S(93.7 vs 149.2 kJ mol-1)催化剂。
如图4b所示,C2H4脱附路线在动力学上仍比CHCH3氢化路线更容易发生;即C2H4脱附途径在以下六种催化剂中占主导地位:Pd-H1.0、Pd-B0.5、Pd-C0.5、Pd-N、Pd-P和Pd-S催化剂,它们可以促进气态C2H4的形成。

图5. 在393K时Pd-H1.0催化剂上C2H2半加氢反应的吉布斯能曲线以及初始状态、过渡状态和最终状态结构
如图5所示,与Pd-H1.0催化剂上的C2H2+C2H2和C2H2+C2H3路线(105.7 vs 157.6,195.8 kJ mol–1)和Pd-C0.5催化剂上的(174.4vs 175.7,192.7 kJ mol-1)相比,C2H3+C2H3耦合路线在动力学上更容易发生。此外,从相同的中间体C2H3开始,C2H3+C2H3耦合路线在动力学上优于Pd-H1.0催化剂上的C2H4脱附路线(72.6 vs 95.6 kJ mol–1,图5),而与Pd-B0.5(90.1 vs 168.4 kJ mol–1)和Pd-C0.5催化剂(83.2 vs 174.4 kJ mol-1)上的C2H2+C2H2或C2H3+C2H3耦合路线相比,C2H4脱附路线在动力学上更容易。因此,掺杂1.0ML次表面H的Pd催化剂不能抑制生成绿油,而掺杂1/2ML次表面B或C的Pd催化可以抑制绿油生成。
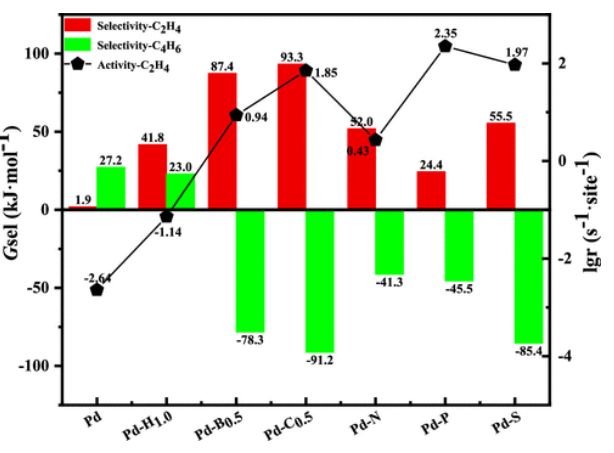
图6. C2H4和C4H6的选择性以及在393K下C2H4在亚表面杂原子掺杂的Pd催化剂上的活性
如图6所示,C2H4选择性(kJ mol–1)的顺序为Pd-C0.5(93.3)>Pd-B0.5(87.4)>Pd-S(55.5)>Pd-N(52.0)>Pd-H1.0(41.8)>Pd-P(24.4)>Pd(1.9)。对于催化剂稳定性,如图6所示,1,3-丁二烯的选择性(kJ mol–1)为Pd-C0.5(−91.2)<Pd-S(−85.4)<Pd-B0.5(−78.3)<Pd-P(−45.5)<Pd-N(−41.3)<Pd-H1.0(23.0)<Pd(27.2)。因此,Pd和Pd-H1.0催化剂均不能抑制绿油生成;然而,将具有1/2ML的B和C原子以及具有1/16 ML的N、P或S原子掺入Pd压面表层可以显著抑制绿油的生成,从而表现出优异的催化剂稳定性。

图7. 亚表面杂原子掺杂钯催化剂的差分电荷密度
对于亚表面杂原子掺杂的钯催化剂,亚表面的H、C、N或S杂原子可以接受来自表面钯原子的电子,而B或P杂原子向表面钯原子提供电子。如图7所示,其电负性顺序为N(3.04)>S(2.58)>C(2.55)>H(2.20)>P(2.19)>B(2.04)。与Pd催化剂相比,随着同一杂原子的覆盖率增加,更多的电荷在杂原子和表面Pd原子之间转移,这在调节催化性能方面起着关键作用。

图8. 亚表面杂原子掺杂的Pd和Pd催化剂上表面Pd原子的d轨道的投影态密度(pDOS)图
如图8所示,Pd-H、Pd-H0.5和Pd-H1.0催化剂的最顶层Pd原子d带中心为−2.01, −2.20和-2.45 eV;与Pd相比(−1.98eV),Pd-H和Pd-H0.5催化剂上的d带中心没有显著变化,而Pd-H1.0催化剂的d带中心变化明显。
因此,更高的电荷转移和更低d带中心的Pd-H1.0催化剂可以降低C2H4吸附能,有利于C2H2氢化,从而提高了Pd-H1.0催化剂上的C2H4活性。对于B原子或C原子掺杂的Pd催化剂,当亚表面B或C原子的覆盖率为1/16 ML时,表面Pd原子的平均Bader电荷变化值仅为0.013和0.013 e,Pd-B和Pd-C催化剂的最顶层Pd原子d带中心变化很小(图8)。Pd-B和Pd-C催化剂上的C2H4吸附构型仍然是稳定的“di-σ”模式。
另一方面,1/16 ML B原子或C原子亚表面掺杂的Pd催化剂表面几乎没有几何结构变化(图1)。因此,1/16 ML B原子或C原子亚表面掺杂对C2H4吸附和C2H4氢化几乎没有影响,则Pd-B和Pd-C催化剂上的C2H4选择性没有明显变化。如图8所示,在Pd-N、Pd-P和Pd-S催化剂上,虽然最顶层Pd原子的d带中心的变化值约为0.09、0.08和0.07eV,但其C2H4活性明显增加。
结论与展望
研究发现,亚表面杂原子的类型和覆盖率可以显著影响C2H2半加氢的催化性能。作者筛选了具有1/2单层(ML)杂原子覆盖率的Pd-B0.5和Pd-C0.5催化剂,以及具有1/16 ML杂原子覆盖度的Pd-N、Pd-P和Pd-S催化剂,它们不仅可以显著提高C2H4的选择性和活性,而且有效地抑制了绿油生成。
亚表面杂原子掺杂可以产生独特的表面Pd电子和几何结构,从而调节催化性能。如在Pd-B0.5和Pd-C0.5催化剂中,Pd表面电子和几何效应在调节活性方面主导作用,而几何效应在Pd-N、Pd-P和Pd-S催化剂中起关键作用。这些发现为设计炔半加氢中的高性能金属催化剂提供了理论指导。
文献信息
Wu Y, Zhao W, Wang Y, et al. Enhancing Catalytic Performance through Subsurface Chemistry: The Case of C2H2 Semihydrogenation over Pd Catalysts[J]. ACS Applied Materials & Interfaces, 2022.
